Актуальность . Пренатальная диагностика (ПД) врождённых пороков развития плода является исключительно важной составляющей дородового наблюдения, позволяя предотвратить рождение детей с тяжёлыми, некорректируемыми пороками развития, с социально значимыми смертельными генными и хромосомными заболеваниями. Это основной фактор снижения заболеваемости, инвалидизации и смертности. К тому же пренатальное выявление врождённых аномалий головного мозга у плода влияет на акушерскую тактику и уменьшает отрицательные психологические и социальные последствия для матери и для семьи в целом, так как врождённые пороки развития (ВПР) головного мозга - это, в основном, тяжёлые и неизлечимые заболевания (ранняя и качественная диагностика пороков развития ЦНС может объективно прогнозировать ситуацию и способствовать правильному выбору тактики лечения). Таким образом, грамотная пренатальная диагностика в сочетании со сбором полной информации, направленной на выявление различных факторов риска, позволяет врачу и беременной повышенного риска адекватно подойти к вопросу о рождении ребенка.
Методы , применяемые для антенатальной (дородовой) диагностики, целесообразно разделить на непрямые, когда объектом исследования является беременная женщина, и прямые, когда исследуется сам плод. Последние могут быть инвазивными и неинвазивными.
Непрямые методы включают изучение акушерского и гинекологического анамнеза, проведение медико-генетического консультирования, а также проведение бактериологических и серологических исследований. Отдельно следует сказать о проведении биохимических скрининговых тестов, направленных на оценку уровней фетопротеина, эстриола, хорионического гонадотропина и др. Следует отметить, что главным назначением непрямых методов является отбор женщин из групп высокого риска для дальнейшего углублённого наблюдения уже на уровне центров репродуктивного здоровья. Основные показания для направления беременной на ПД во всём мире примерно одинаковы. Они включают:
[1
] возраст женщины старше 35 лет;
[2
] наличие не менее двух самопроизвольных выкидышей (абортов) на ранних сроках беременности;
[3
] наличие в семье ребёнка или плода от предыдущей беременности с болезнью Дауна, другими хромосомными болезнями, с множественными врождёнными пороками, семейное носительство хромосомных перестроек;
[4
] многие моногенные заболевания, ранее диагностированные в семье или у ближайших родственников;
[5
] применение перед и на ранних сроках беременности ряда фармакологических препаратов;
[6
] перенесённые вирусные инфекции (гепатит, краснуха, токсоплазмоз и др.);
[7
] облучение кого-нибудь из супругов до зачатия.
читайте также статью: Токсоплазмоз (на сайт)
Основными неинвазивными методами являются [1 ] ультразвуковое (эхографическое) исследование (УЗИ) и [2 ] магнитно-резонансная томография (МРТ).
Ультразвуковое исследование ЦНС и спинного мозга у плода является одной из самых важных и ответственных задач пренатальной эхографии, поскольку оказывает значительное влияние на оптимизацию акушерской тактики и принятие родителями решения о пролонгировании или прерывании беременности. Оптимальными сроками эхографического обследования у плода для исключения большинства пороков и заболеваний ЦНС являются:
[1
] 11 - 14 недель гестации;
[2
] 19 - 21 неделя гестации;
[3
] 30 - 33 недели гестации.
Это соответствует начальным этапам манифестации различных групп пороков и заболеваний ЦНС, а так же обеспечивает преемственность диагностики и общепринятых стандартов тактики ведения беременности и родов. При этом схема УЗ-обследования плода должна включать оценку эхографической анатомии плода с изучением костей свода черепа, основных структур головного мозга, воротникового пространства, профиля, орбит, носовых костей, ориентации сердца, позвоночника, передней брюшной стенки, желудка, кишечника, мочевого пузыря и конечностей.
Данные, полученные рядом авторов, свидетельствуют о высокой диагностической ценности УЗИ в пренатальной диагностике ВПР, позволяющей выявить более 80 - 90% пороков развития плода во время беременности. Значительная часть грубой патологии развития плода может быть выявлена в I триместре при соблюдении сроков обследования и всех требований скринингового протокола оценки анатомии плода.
Использование современных УЗ-аппаратов экспертного класса, трансвагинальной эхографии с применением 3/4D режимов значительно увеличивает точность диагностики. Исследование головного мозга плода в средне-сагиттальной плоскости, получение которой практически стало возможным в большинстве случаев с использованием трёхмерной эхографии, позволяет дифференцированно подходить к оценке нормы и патологии срединных структур мозга. Аксиальные сечения головного мозга плода, используемые в рутинной УЗ-практике, не позволяли получить ясную картину мозолистого тела. Расширение протокола первого скринингового исследования оценкой «интракраниальной прозрачности» повысило точность диагностики spina bifida в I триместре беременности.
Использование цветового допплеровского картирования для визуализации сосудов головного мозга у плода в раннем онтогенезе достоверно позволяет визуализировать сосуд, кровоснабжающий конкретную структуру, на 2 - 5 недель ранее их стандартного эхографического выявления уже в конце I и в начале II триместра беременности. Комплексная оценка развития борозд и субарахноидальных пространств даёт возможность диагностировать нарушение формирования коры головного мозга уже во II триместре беременности. Аномалии развития ЦНС, как нарушения эмбриогенеза и раннего плодового органогенеза, могут быть выявлены с использованием современной эхографии до 21 недели гестации. В то же время, эхографические признаки деструктивных повреждений и объёмных образований ЦНС могут быть диагностированы только во II и III триместрах беременности (С.М. Воеводин, 2012).
Трансвагинальная эхография с изучением анатомических структур плода при УЗИ в 11 - 14 недель беременности является высокоинформативным методом пренатальной диагностики в ранние сроки беременности, позволяющим обнаружить более половины всех врождённых пороков, являющихся в большинстве случаев грубыми.
Необходимо также отметить ценность для диагностики ВПР такого ультразвукового признака, как многоводие. Степень тяжести многоводия коррелирует с частотой врождённых пороков развития плода. Установлена прямая связь между количеством околоплодных вод и частотой развития врождённых пороков развития плода.
В целом успех диагностики зависит также от вида ВПР (ряд аномалий достаточно сложен для диагностики), срока беременности, в котором проводится исследование, количества околоплодных вод, конституциональных особенностей пациентки (выраженное ожирение создаёт сложности при трансабдоминальном сканировании). Другими фактрами, затрудняющими правильную интерпретацию результатов скрининга, могут быть гестационный возраст, многоплодная беременность, этническая принадлежность родителей, диабет у матери. Достаточно сложна пренатальная диагностика агенезии мозолистого тела (основные сроки выявления которой - II - III триместры беременности), спинномозговой грыжи. В то же время, несмотря на высокую точность и относительную простоту УЗ-диагностики, таких абсолютно летальных пороков, как акрания и анэнцефалия, некоторые из них выявляются после 24 недель беременности, что может свидетельствовать о недостаточно высокой квалификации врачей, несоблюдении скрининговых сроков и методики обследования, позднем направлении женщин на второй-третий уровень обследования. Ещё одним из негативных факторов может быть отказ женщин от пренатального кариотипирования. Кроме этого, остаются до конца нерешёнными такие вопросы пренатальной УЗ-диагностики ВПР ЦНС, как проведение дифференциальной диагностики при редко встречающихся пороках.
МРТ является высокоинформативным методом пренатальной диагностики и может быть использована при заподозренных пороках плода в ходе УЗисследования, особенно в случаях аномалий ЦНС. Во II и III триместрах беременности использование в качестве дополнительного обследования МРТ улучшает диагностику пороков ЦНС и даёт возможность уточнить диагноз при патологии коры больших полушарий, срединных структур головного мозга, задней черепной ямки и нарушениях ликворо-динамики. МРТ с успехом может быть использована в тех случаях, когда результаты УЗИ являются недостаточно информативными. Однако, несмотря на то, что МРТ способна подтвердить диагноз, установленный на УЗИ, и получить более подробные данные, относительно высокая стоимость, отсутствие стандартизированных реферативных значений и ограниченная доступность МРТ, являются причиной того, что УЗИ остаётся исследованием выбора диагностики ВПР плода.
о том безопасно ли МРТ во время беременности Вы можете прочитать
Исследованию маркёрных эмбриональных белков в сыворотке крови матери. В последние годы особенно важная роль принадлежит исследованию маркёрных эмбриональных белков в сыворотке крови матери, таких, как альфа-фетопротеин (АФП), хориальный гонадотропин (ХГЧ), свободный эстрадиол и некоторые другие. Целью таких исследований является выявление женщин с высоким риском рождения детей с врождёнными и наследственными пороками. Изменения сывороточных маркёров характерны для 90,9 % женщин с врождёнными пороками развития ЦНС. Проведённое в оптимальные сроки (15 - 16-недельной беременности) с использованием трёх тест-систем исследование позволяет выявить до 80% плодов с дефектами развития внутренних органов и до 65% - с хромосомными болезнями.
Пренатальная лабораторная диагностика аномалий нервной трубки базируется преимущественно на определении уровня фетального АФП. Этот протеин является основным компонентом белковой системы сыворотки эмбриона и определяется на 30-й день гестации. Повышение уровня АФП в амниотической жидкости является признаком наличия открытого дефекта нервной трубки. Во II триместре беременности при УЗИ можно достоверно выявить аномалию головного мозга плода. Поскольку в образовании эстриола принимают участие как плод, так и плацента, уровень эстриола может служить идеальным показателем функционирования фетоплацентарной системы. Чем ниже уровень гормона, тем выше вероятность развития патологического состояния плода.
В то же время, интерпретация изолированных результатов биохимического тестирования может быть затруднена. При рассмотрении кривых распределения значений основных маркёров наблюдается большая зона перекрывания нормы и патологии, что не позволяет использовать только один маркёр, необходим полный их комплекс: ХГЧ, АФП и эстриол.
Биохимическое тестирование в I триместре гестации, которое включает [1 ] определение концентраций прогестерона, [2 ] неконъюгированного эстриола, [3 ] β-фракции хорионического гонадотропина (β-ХГЧ) и [4 ] протеина, ассоциированного с беременностью (7 - 8 или 11 - 12 недель), является более эффективным методом пренатального скрининга, чем традиционный «тройной» тест II триместра, т.е. АФП, β-ХГЧ, эстриол - в 16 - 17 недель беременности.
Несмотря на достаточно высокую эффективность современных неинвазивных методик, достаточно полная информация о кариотипе зародыша, биохимических и генотипических особенностях его клеток может быть получена только на основании соответствующих исследований непосредственно тканей самого плода или его провизорных органов (плаценты, хориона), полученных инвазивным путём на любом сроке беременности. Наиболее часто проводится амниоцентез для выявления хромосомных аномалий и генетических мутаций, однако амниотическая жидкость может быть также использована для диагностики дефектов нервной трубки. Наиболее частыми возможными рисками процедуры являются спонтанный аборт (от 0,5% до 1,0%), кровянистые выделения после процедуры, инфекция, разрыв плодных оболочек и повреждения или потеря плода.
Наиболее информативным является комплексное обследование беременных с использованием современных УЗ-технологий в сочетании с биохимическим скринингом, что повышает точность диагностики ВПР ЦНС плода на ранних сроках II триместра. Кроме того, выявление признаков гипоксии плода может свидетельствовать о возможном наличии не только фетоплацентарной недостаточности, но и врождённой патологии, так как гипоксия и задержка внутриутробного развития значительно ухудшают прогноз при ВПР, совместимых с жизнью.
Следует помнить , что врождённые аномалии головного мозга, выявленные пренатально или же в неонатальном периоде, не считая непостоянных внешних пороков развития, [!!! ] могут НЕ иметь каких-либо специфических клинических симптомов:
[1 ] при нейросонографическом и магнитно-резонансном исследованиях, помимо конкретных нозологических форм врождённых пороков развития головного мозга (синдром Киари, синдром Денди-Уокера, окклюзионная гидроцефалия и др.), следует обращать внимание на гипоплазии, которые встречаются значительно чаще;
[2 ] при отсутствии пренатальной и неонатальной диагностики с использованием УЗИ и МРТ появляются условия для поздней диагностики, в тот период, когда в клинической картине на первое место выдвигаются психоневрологические симптомы, при этом наличие умственной отсталости, внутричерепной гипертензии может служить основанием для постановки таких диагнозов как ДЦП, гидроцефалия и др.
Министерство образования и науки Российской Федерации
Федеральное агентство по образованию
ГОУ ВПО "Алтайский Государственный Университет"
Факультет психологии и философии
Кафедра общей и прикладной психологии
Тема: Пороки развития головного мозга
Контрольная работа по предмету: Анатомия центральной нервной
системы человека
Выполнила:
студентка гр.1881 в
заочного отделения ФПФ
Шмакова Ольга Сергеевна
Проверила:
к. м. н., доцент
Гречишникова Людмила Михайловна
Введение
2.1 Врожденная гидроцефалия
2.2 Краниостеноз
2.3 Агенезия мозолистого тела
2.4 Передние грыжи
Библиография
Введение
Аномалии развития нервной системы можно условно разделить на сочетающиеся с распознаваемыми соматическими пороками и на ограниченные лишь пределами нервной системы (поражение нервной системы имеет место при 60% от общего числа). Целесообразна также классификация аномалий развития и врожденных пороков на группу, обусловленную приобретенными или внешними факторами, и на генетически детерминированную группу. Однако в некоторых случаях в их основе лежит сложное взаимодействие генетических факторов и условий окружающей среды.
Факторы, влияющие на развитие нервной системы:
Неблагоприятное влияние каких-либо факторов на мозг в период его развитие представляет собой сложную производную от степени тяжести повреждения, его длительности, специфического биологического влияния вредоносного агента и определенной стадии развития, во время которого это воздействие оказывается. Особенно важно знать причины, вызвавшие аномалии, связанные с воздействиями окружающей среды, поскольку их можно устранить.
Токсины, присутствующие в организме матери, могут быть причиной, вызывающей повреждение развивающегося мозга и нервов. Алкогольный синдром плода. существенная причина задержек психического развития, обусловлен воздействием на плод избыточных количеств алкоголя, потребляемых матерью. Кроме того, на формирование мозга у плода может повлиять применение матерью медикаментозных средств, в особенности антиконвульсантов. Грубые аномалии у плода дает триметадион. Установлено, что вальпроевая кислота может привести к образованию spinabfidа. Прием матерью фенитоина в первые месяцы беременности вызывает незначительное, но четко распознаваемое влияние на формирование мозга и соматическое развитие. Изотретиноин, препарат, применяемый при акне, вызывает врожденные пороки мозга. Установлено, что дефекты развития мозга у плодов в Минимата Бей, Япония, обусловливались воздействием органического ртутного токсина. Возникновение микроцефалии и умственную отсталость могут обусловливать радиация я радиомиметические факторы, воздействующие на женщину в I триместре беременности.
I. Классификация пороков развития внутренних органов
Под термином "врожденный порок развития" следует понимать стойкие морфологические изменения органа или всего организма, выходящие за пределы вариаций их строения.
Врожденные пороки развития возникают внутриутробно в результате нарушения процессов развития зародыша или (много реже) после рождения ребенка, как следствие нарушения дальнейшего формирования органов.
Как синонимы термина "врожденные пороки развития" могут применяться термины "врожденные аномалии", "врожденные пороки" и "пороки развития", "аномалии развития".
Понятие "врожденный порок" не ограничивается нарушениями развития, а включают в себя и врожденные нарушения обмена веществ.
К врожденным порокам не следует относить постнатальные нарушения пропорций или размеров органов, являющиеся проявлением эндокринных расстройств (гипофизарная карликовость, гигантизм, акромегалия).
Все пороки развития внутренних органов можно подразделить на 4 группы:
Аномалии количества:
А) Отсутствие органа, связанное с агенезией или аплазией.
1) Агенезия - неразвитие органа, зависящее от отсутствия его закладки у эмбриона.
2) Аплазия - неразвитие эмбрионального зачатка, выражается, как и агенезия, во врожденном отсутствии органа.
Б) Удвоение органа (дупликация) или образование добавочных органов - обусловлено множественной эмбриональной закладкой или разделением зачатка органа.
В) Слияние (неразделение) органов.
Аномалии положения:
А) Гетеротопия - закладка органа у зародыша в необычном месте, в котором и происходит его дальнейшее развитие.
Б) Дистопия - смещение органа в необычное место в эмбриональном периоде.
В) Инверсия - обратное положение органа относительно его собственной оси или срединной плоскости тела вследствие нарушения эмбрионального поворота.
Аномалии формы и размера:
А) Гипоплазия - недостаточное развитие органа вследствие задержки на какой-либо стадии эмбриогенеза, проявляющееся дефицитом относительной массы или размеров органа, превышающим отклонение в две сигмы от средних показателей для данного возраста. Гипопластический орган уменьшен в размерах, функция его понижена или совсем отсутствует.
1) Простая гипоплазия - не сопровождается нарушением структуры органов.
2) Диспластическая гипоплазия - сопровождается нарушением структуры органов.
Б) Гиперплазия (гипертрофия) - увеличение относительной массы или размеров органа за счет увеличения количества (гиперплазия) или объема (гипертрофия) клеток.
В) Сращение парных органов - зависит от слияния их закладок в эмбриональном периоде.
Аномалии строения (структуры):
А) Атрезия - полное отсутствие канала или естественного отверстия тела.
Б) Гетероплазия - нарушение дифференцировки отдельных типов тканей.
В) Дивертикул - аномальный вырост полых органов.
Г) Дисплазия - нарушение формирования составных тканевых элементов органа.
Д) Стеноз - сужение канала или отверстия.
Е) Гамартия - неправильное соотношение тканей в анатомических структурах или наличие отсутствующих в норме остатков зародышевых образований в зрелом организме.
Ж) Киста дизонтогенетическая.
Кроме того, может наблюдаться абиотрофия - скрытая аномалия органа или системы организма, характеризующаяся резким снижением адаптационных возможностей и проявляющаяся преждевременным ослаблением функции при обычном уровне деятельности.
По этиологическому признаку различают 3 группы пороков:
Наследственные - пороки, возникшие в результате мутаций, то есть стойких изменений наследственных структур в половых клетках (гаметах) - гаметические мутации, или в зиготе зиготические мутации.
Экзогенные - пороки, обусловленные повреждением тератогенными факторами непосредственно эмбриона или плода. Поскольку пороки развития, вызванные тератогенами, могут копировать генетически детерминированные пороки развития, их нередко называют фенокопиями.
Мультифакториальные - пороки, которые произошли от совместного воздействия генетических и экзогенных факторов, причем ни один из них отдельно не является причиной порока.
В зависимости от объекта воздействия вредящих факторов врожденные пороки могут быть разделены на пороки, возникшие в результате:
1) гаметопатий,
2) бластопатий,
3) эмбриопатий,
4) фетопатий.
1. Гаметопатии - поражения половых клеток, "гамет".
2. Бластопатии - поражения бластоцисты, то есть зародыша первых 15 дней после оплодотворения (до момента завершения дифференциации зародышевых листков и начала маточно-плацентарного кровообращения).
3. Эмбриопатии - пороки, возникшие в результате повреждения эмбриона независимо от этиологии в период от 16-го дня после оплодотворения до конца 8-й недели.
4. Фетопатии - повреждения плода в период от 9-й недели до окончания родов. Пороки этой группы сравнительно редки.
По распространенности в организме врожденные пороки развития подразделяют на 3 группы:
1. Изолированные - локализованные в одном органе.
2. Системные - в пределах одной системы органов.
3. Множественные - локализованные в органах двух и более систем.
Врожденные пороки ЦНС по частоте занимают первое место среди других пороков, встречаются в 30% случаев среди пороков развития, обнаруживаемых у детей.
Этиология и патогенез. Из экзогенных факторов точно установлено значение вируса краснухи, иммунодефицита человека, простого герпеса, предполагается влияние вирусов цитомегалии, Коксаки, лекарственных препаратов (хинин, гидантоин и др.), алкоголя, лучевой энергии, гипоксии. Несомненное значение имеют генные мутации; при хромосомных болезнях в числе множественных пороков они встречаются почти как правило. Развитие порока связано с воздействием повреждающего агента в течение всего эмбрионального периода, включая ранний фетальный. Наиболее тяжелые пороки возникают при повреждении в начале закладки нервной трубки (3-4-я неделя внутриутробной жизни.
II. Пороки развития головного мозга
Головной мозг (encephalon) - передний отдел центральной нервной с окружающими его оболочками расположен в полости мозгового отдела черепа. Верхняя выпуклая поверхность головного мозга соответствует своей формой внутренней поверхности свода черепа, а нижняя, более плоская, со сложным рельефом, - внутреннему основанию черепа.
Масса головного мозга взрослого человека колеблется от 1100 до 2000 г; у мужчин в среднем она составляет около 1394 г, а у женщин 1245 г. После 60 лет масса и объем мозга несколько уменьшаются.
Наиболее частыми причинами различных пороков развития головного мозга являются неправильная закладка нервной системы или поражение ее в период эмбрионального развития вследствие изменений генетической информации (нарушения гистогенеза и цитоархитектоники головного мозга) или влияние внешних факторов., некоторых инфекций, перенесенных матерью в период беременности (токсоплазмоз, краснуха, цитомегалия, вирусный гепатит), воздействия ионизирующего излучения, травм, а также в результате вредного воздействия некоторых химических веществ. Пороки развития головного мозга (кроме мозговых грыж), как правило, сопровождаются олигофренией.
К основным порокам развития головного мозга относятся пороки развития системы желудочков головного мозга и коры большого мозга, агенезии, краниосхизы.
Пороками развития системы желудочков являются:
порэнцефалия - появление кист различных размеров в головном мозге, сообщающихся с боковыми желудочками мозга, выстланных эпендимой. От истинной порэнцефалии следует отличать ложную, при которой кисты не сообщаются с путями оттока ликвора и образуются на месте бывших размягчений ткани головного мозга.
врожденная гидроцефалия - увеличение желудочков головного мозга. Гидроцефалия в результате пороков развития ц. н. с. проявляется в форме гидроаэнцефалии (сочетание врожденной гидроцефалии с выраженной атрофией больших полушарий головного мозга), гидромезэнцефалии.
Гидроцефалией, или водянкой головного мозга, называют патологическое состояние, характеризующееся увеличением количества жидкости в полости черепа.
Различают:
а) общую гидроцефалию с увеличением содержания жидкости в желудочках мозга и субарахноидальном пространстве;
б) внутреннюю, или желудочковую форму, при которой имеется избыточное содержание жидкости внутри желудочков;
в) редко наблюдающуюся наружную гидроцефалию с избыточным содержанием жидкости в субарахноидальном пространстве при нормальном содержании её в желудочках, которое развивается ex vacuo при атрофии мозга.
Общее количество жидкости, циркулирующее в норме в желудочках и субарахноидальном пространстве головного и спинного мозга, ровняется в среднем 150 мл, из которых одна половина распределяется примерно поровну между желудочками и субарахноидальным пространством головного мозга, а вторая - находится в субарахноидальном пространстве спинного мозга.
В последние десятилетия считается общепринятым, что ликвор или спинномозговая жидкость вырабатывается в основном сосудистыми сплетениями, располагающимися в боковых, III и IV желудочках мозга, циркулирует по желудочковой системе мозга в направлении боковые желудочки à отверстия Монро à III желудочек à сильвиев водопровод à IV желудочек и через отверстия Мажанди и Люшка оттекает в субарахноидальное пространство. Основная масса спинномозговой жидкости резорбируется на наружной поверхности больших полушарий.
С нейрохирургической точки зрения наиболее важно деление гидроцефалии на следующие две основные формы:
открытую (сообщающуюся) форму, являющуюся следствием повышения продукции ликвора (гиперсекреторная форма) или замедления всасываемости его (арезорбтивная форма), или сочетания обоих факторов. При этом циркуляция ликвора между желудочковой системой и субарахноидальным пространством не затруднена;
закрытую (окклюзионную) внутреннюю форму воспалительной, опухолевой или другой этиологии, при которой имеется препятствие на пути оттока ликвора из желудочковой системы в базальные цистерны и субарахноидальное пространство.
По месту обтурирующего процесса, который обычно локализуется в наиболее узких местах коммуникаций, различают следующие виды закрытой гидроцефалии:
а) с уровнем окклюзии у отверстия Монро;
б) у сильвиева водопровода;
в) у отверстий Мажанди и Люшка.
По времени развития различают гидроцефалию врожденную и приобретенную (симптоматическую), которые могут быть как сообщающимися, так и окклюзионными.
2.1 Врожденная гидроцефалия
Эта форма представляет собой нарастающую внутричерепную водянку, вызванную причинами, действие которых на мозг относится к периоду внутриутробного развития или родов. Этими причинами являются: а) токсоплазмоз и другие инфекционно-воспалительные заболевания матери, вызывающие у плода менингоэнцефалит или задержку развития структур мозга, обеспечивающих всасывание ликвора; б) внутричерепная травма в родах с возникновением внутричерепных кровоизлияний.
Различают две основные формы врожденной гидроцефалии:
а) сообщающуюся;
б) окклюзионную.
Развиваются они в тех случаях, когда отсутствует образование отверстий Мажанди и Люшка на 3-4 месяце внутриутробного развития или когда эти отверстия закрываются в более позднем периоде развития вследствие воспалительных процессов в мозге плода. В последних случаях закупорка может произойти как на уровне указанных отверстий, так и сильвиева водопровода. Количество сообщающихся форм значительно преобладает над окклюзионными. По данным В.П. Пурина (1968), среди 227 детей с врожденной гидроцефалией у 180 была сообщающаяся и у 47 окклюзионная форма.
Врожденные гидроцефалии могут осложняться энцефаломаляцией. Большей частью в белом веществе. В результате развивается та или иная степень атрофии мозга, иногда достигающая такой степени выраженности, что вместо полушарий головного мозга имеются тонкостенные пузыри, заполненные ликвором.
Клиническая картина врожденной гидроцефалии проявляется увеличением и характерным изменением формы черепа, иногда очень значительным, что во многих случаях проявляется сразу после рождения. В дальнейшем под влиянием нарастающего давления ликвора это увеличение быстро прогрессирует, происходит истончение костей черепа, расширение черепных швов и повышение напряжения; пульсация родничков отсутствует. Так как лицевой скелет при этом не увеличивается, лицо приобретает при этом треугольную форму и по сравнению с большой шарообразной головой кажется маленьким; оно бледно, морщинисто и старообразно.
Многообразная неврологическая симптоматика является следствием повышения внутричерепного давления с развитием атрофических и дегенеративных процессов в мозге и черепно-мозговых нервах. Стойкое повышение внутричерепного давления ведет к сдавлению капилляров мозга и как следствие - к атрофии нервной ткани. Поражение черепно-мозговых нервов проявляется в первую очередь нарушением функции зрения и различной степени атрофии зрительных нервов, снижением зрения иногда с исходом в слепоту. Нарушение двигательных функций проявляется в том, что дети поздно начинают сидеть и ходить и плохо удерживают голову. Может отмечаться значительное отставание в интеллектуальном развитии, нередко имеют место повышенная возбудимость и раздражительность или вялость и адинамия, безучастность к окружающему. Парезы и параличи конечностей выражены в разной степени. Отсталость в умственном развитии колеблется в широких пределах. Нередко наблюдается слабоумие и идиотизм.
Наряду с общим отставанием в психическом развитии у некоторых больных гидроцефалией наблюдается хорошая сохранность, и даже высокое развитие отдельных психических функций: необычайная механическая память, способность к счету, музыкальная одаренность.
Обострение гидроцефального синдрома с развитием острых окклюзионных приступов проявляется быстрым развитием тяжелого состояния с резко выраженными головными болями, рвотой, головокружением, брадикардией, которая может сменится тахикардией, тоническими судорогами, бессознательным состоянием и летальным исходом.
Развитие заболевания при врожденной форме гидроцефалии может в любой стадии спонтанно приостановиться. При этом в легких случаях может наблюдаться и полное практическое выздоровление. В тяжелых, прогрессирующих случаях при отсутствии своевременного оперативного вмешательства прогноз врожденной гидроцефалии неблагоприятен: большинство детей умирает в первые месяцы или годы жизни от различных интеркуррентных заболеваний и осложнений (пролежни, дистрофии и т.д.) и только немногие доживают до старшего возраста.
При решении вопроса о показаниях к оперативному вмешательству в случаях врожденной гидроцефалии у детей следует учесть два основных вопроса:
1) прогрессирует ли увеличение объема головки, либо при стабилизации патологического процесса в случаях сообщающейся гидроцефалии отсутствуют показания к оперативному вмешательству;
2) имеется ли открытая либо закрытая формы гидроцефалии, т.к при окклюзионной форме имеются показания для оперативного вмешательства во всех случаях (при отсутствии противопоказаний).
Первый вопрос решается динамическим наблюдением за ребенком с измерением различных размеров его головки, второй на основании анализа клинических данных и пневмографии. Если при пневмоэнцефалографии воздух проникает в желудочковую систему, имеется сообщающаяся форма гидроцефалии, если нет, - вероятно, окклюзионная. В сомнительных случаях диагноз уточняется с помощью вентрикулографии, которая может быть выполнена у маленьких детей путем пункции через роднички.
Оперативное вмешательство целесообразно предпринять относительно рано, когда еще не развились необратимые изменения в мозге и организме, - в 6-мес или годовалом возрасте. При окклюзии сильвиева водопровода, возникающей вследствие родовой травмы, хирургическое вмешательство считается показанным уже в первые недели жизни, т.к консервативная терапия в этих случаях неэффективна. В настоящее время при врожденной гидроцефалии применяются в основном универсальные методы оперативного вмешательства. Противопоказано оперативное лечение в далеко зашедших стадиях поражения мозга с развитием резко выраженной атрофии мозговой ткани, при необратимых изменениях интеллекта, слепоте.
На основании течения заболевания у 227 детей с врожденной гидроцефалией В.П. Пурин приходит к заключению, что в 44% случаев процесс завершается спонтанной компенсацией нарушений ликвородинамики без грубых нарушений психики и моторики ребенка; в 30% только своевременное радикальное хирургическое вмешательство может позволить надеяться на благоприятный исход заболеваний, а в 20% случаев прогнозов оказывается безнадежным вследствие грубых изменений мозга, имеющихся к моменту рождения.
К порокам развития коры большого мозга (обычно сочетаются с недостаточной дифференцированностью ее клеток) относятся:
макрогирия (увеличение размеров извилин большого мозга при уменьшении их числа),
микрогирия (уменьшение размеров извилин большого мозга при увеличении их числа),
агирия - отсутствие извилин большого мозга (нередко сочетается с порэнцефалией).
Эта группа пороков клинически проявляется слабоумием, спастическими парезами, судорожными припадками.
Микрогирия и агирия могут сочетаться с пороком развития черепа - краниостенозом.
2.2 Краниостеноз
Краниостеноз - преждевременное закрытие черепных швов, ведущее к ограничению объема черепа, его деформации и повышению внутричерепного давления. Вследствие преждевременного заращения одного или нескольких швов черепа ограничивается рост костей в направлении, перпендикулярном закрывшемуся шву. Компенсаторный рост черепа происходит за счет сохранившихся швов, что неизбежно ведет к его деформации. В одних случаях преждевременное заращение швов ограничивается лишь изменением формы головы, в других - компенсаторный рост костей черепа не успевает удовлетворить требованиям растущего мозга в объеме, что ведет к прогрессивному повышению внутричерепного давления. Первоначально недостающий объем черепа до определенного предела уравновешивается внутричерепными компенсаторными механизмами, в основном за счет перераспределения спинномозговой жидкости в ликворных, пространствах головного и спинного мозга, изменения процессов продукции ликвора, перераспределения кровотока в полости черепа и истончения костей черепа. Любое заболевание, сопровождающееся даже незначительным отеком головного мозга (инфекция, травма черепа и пр), иногда приводит к истощению внутричерепных компенсаторных механизмов, прогрессирующему повышению внутричерепного давления, нарушению крово- и ликвороциркуляции в полости черепа, т.е. к декомпенсации заболевания.
По степени клинического проявления заболевания различают краниостеноз компенсированный и декомпенсированный.
По динамике заболевания различают краниостеноз прогрессирующий и стабилизировавшийся.
В практическом отношении установить диагноз краниостеноза, который определял бы сущность процесса и необходимое лечение. В связи с этим различают формы краниостеноза в зависимости от вида и количества преждевременно закрывшихся черепных швов: краниостеноза с заращиванием только коронарного и сагиттального швов или общий краниостеноз, когда преждевременно окостеневают все черепные швы.
Наиболее часто встречается изолированное заращение сагиттального и сочетанное закрытие сагиттального и коронарного швов.
Распознавание краниостеноза в детском возрасте не представляет затруднений. Первым наглядным его признаком является изменение формы головы. Чем раньше и быстрее закрываются черепные швы, тем скорее меняются формы и размеры черепа и тем раньше проявляется заболевание. В большинстве случаев форма головы настолько показательна, что уже по одному внешнему виду можно определить форму краниостеноза.
Преждевременное заращение коронарного шва ограничивает рост черепа в переднезаднем направлении. Компенсаторно увеличивается высота головы, преимущественно за счет значительного выбухания области лобного родничка.
Закрытие сагиттального шва ведет к уменьшению поперечного размера черепа, уплощению теменных костей, которые соединяются почти под острым углом, образуя по средней линии костный гребень. Избыточный рост костей у сохранившихся поперечных швов значительно увеличивает продольный диаметр.
В случае преждевременного закрытия всех швов уменьшаются продольный и поперечный диаметры, чрезмерно увеличивается высота черепа, который суживается кверху и приобретает остроконечную форму.
Неврологическая симптоматика при краниостенозе характеризуется общемозговыми симптомами, обусловленными повышением внутричерепным давлением и затруднением венозного оттока из полости черепа. В стадии декомпенсации отмечается значительное повышение внутричерепного давления. Ребенок, родившийся с закрытыми черепными швами и измененной формой головы, обычно беспокоен, плаксив, плохо спит по ночам, отказывается от груди. Иногда отмечается неукротимая рвота, которая нередко расценивается, как кишечная интоксикация. В более старшем возрасте повышение внутричерепного давления вызывает прогрессивно нарастающие приступообразные головные боли, к ним присоединяется тошнота, рвота, не связанная с приемом пищи.
Нарушения психики связаны с внутричерепной гипертензией, сводятся к раздражительности или заторможенности, снижению памяти и внимания. В раннем возрасте отмечается задержка умственного развития без явных клинических признаков повышения внутричерепного давления. Особенностью течения краниостеноза можно считать появление у ряда больных судорожных припадков. Понижение остроты зрения обычно развивается в раннем детском возрасте и реже после 7 лет.
Лечение хирургическое, направлено на увеличение объема черепа.
При агенезиях головного мозга могут отсутствовать или быть недоразвитыми различные его отделы: лобные, теменные, височные, реже затылочные доли, мозолистое тело, мозжечок, мозговой ствол. Агенезии головного мозга часто сочетаются с дефектами развития черепных нервов, спинного мозга, дисплазией внутренних органов.
2.3 Агенезия мозолистого тела
Мозолистое тело представляет собой самую большую спайку мозга, объединяющую полушария между собой. Данная структура имеет важное значение в координации информации и обмене сенсорными стимулами между полушариями, в процессе обучения и памяти.
Агенезия мозолистого тела может встречаться изолированно или в сочетании с другими пороками развития головного мозга. J. S. Jeret et al. проанализировали 705 случаев агенезии мозолистого тела (включая данные литературы и свои собственные наблюдения) и пришли к выводу, что данный порок в 23% сочетается с гидроцефалией, в 15% - с микроцефалией, в 23% - с гетеротопии ей серого вещества мозга полимикрогириеи, в 6% - с микрогирией, в 2,4% - с пахигирией, в 3% - с агирией и в 3% - с аринэнцефалией. В 23% случаев агенезии мозолистого тела сочетались с кистами различной локализации (межполушарными, порэнцефалическими, арахноидальными, синдромом Денди-Уокера). Опухоли, включающие липому, папиллому, менингиому, были отмечены в 7% всех случаев. В 20% наблюдались гипертелоризм и другие лицевые аномалии. Часто отмечались поражения глаз, включающие ретинальные лакуны (синдром Айкарди), колобомы, микрофтальмия и катаракты. В литературе имеются указания на то, что агенезия мозолистого тела нередко наблюдается при синдроме Арнольда-Киари II типа, черепно-мозговых грыжах, хромосомных аномалиях.
Клинически они проявляются выраженной задержкой психического и физического развития, очаговой неврологической симптоматикой (парезами, параличами, судорожными припадками).
В случаях изолированного порока агенезия мозолистого тела может не сопровождаться какой-либо неврологической симптоматикой и обнаруживаться случайно. Степень дефекта мозолистого тела зависит от стадии развития плода. Первичная агенезия обычно развивается до 12 недель жизни плода и часто комбинируется с другими пороками развития мозга. Вторичное поражение является следствием энцефаломаляции в более поздних стадиях развития плода в результате токсических, травматических повреждении, аноксии в системе передних мозговых артерий, внутриутробных воспалительных процессов. Агенезия мозолистого тела может быть тотальной или частичной. В случаях частичной агенезии отсутствует задняя часть мозолистого тела, значительно реже - передняя.
Диагноз устанавливается на основании нейровизуальных методов (нейросонография, Компьютерная томография, магнитно-резонансная томография головного мозга). Самым достоверным обследованием считается МРТ.
Пороки развития головного мозга, сочетающиеся с дефектами лицевого и мозгового черепа, называют краниосхизами. К ним относятся:
ацефалия - (часто сочетается с дефектами развития спинного мозга и внутренних органов);
анэнцефалия - агенезия головного мозга, при которой отсутствуют передние, средние, иногда и задние его отделы. Продолговатый и спинной мозг сохранены. На месте головного мозга обнаруживается соединительная ткань, богатая сосудами, в которой встречаются отдельные нейроны и клетки нейроглии (сочетается с акранией отсутствием костей свода черепа, покрывающих их мягких тканей и кожи);
гемицефалия - частичное недоразвитие различных отделов головного мозга, отсутствие, как правило, крыши черепа, а иногда и кожного покрова.
При этих сочетанных пороках новорожденный нежизнеспособен; при некоторых других видах краниосхиза, например циклопии (редкий порок, характеризующийся наличием одного или двух глазных яблок, расположенных в одной глазнице, с одновременным пороком развития носа и обонятельной доли головного мозга. Назван из-за сходства лица плода с лицом мифического чудовища - циклопа), плод жизнеспособен.
К группе краниосхизов принадлежат врожденные грыжи головного мозга.
Черепно-мозговая грыжа - врожденный порок развития черепа и мозга, характеризующийся выпячиванием мозга и его оболочек через дефект в черепе. Причиной этого нарушения является влияние вредных факторов в раннем онтогенезе.
В основу классификации форм черепно-мозговых грыж положены данные патолого-анатомических исследований. В зависимости от того, что является содержимым черепно-мозговой грыжи, большинство авторов различает несколько основных форм:
Менингоцеле - выбухание через дефект в черепе измененных мягкой и паутинной оболочек с заполнением выпячивания измененной цереброспинальной жидкостью. Твердая мозговая оболочка не принимает участия в образовании грыжевого выпячивания, а прикрепляется к краям дефекта кости со стороны полости черепа.
Энцефалоцеле - помимо мягкой и паутинной оболочек, содержимым грыжевого выпячивания является измененная мозговая ткань.
Энцефалоцистоцеле - в грыжевое выпячивание, кроме оболочек и мозговой ткани, вовлекается часть расширенного желудочка мозга. При передних грыжах - передний рог, при задних - задний рог бокового желудочка.
Выделяют также скрытую форму грыжи головного мозга (дефект костей черепа, но без заметной эктопии содержимого черепа) и отщепленную мозговую грыжу (грыжа не сообщается с полостью черепа).
В зависимости от локализации грыжи головного мозга бывают передними, задними, базальными и сагиттальными.
2.4 Передние грыжи
При передних мозговых грыжах дефект на основании черепа локализуется lamina cribrosa, что является отверстием костного грыжевого канала. Наружное выходное отверстие грыжевого канала располагается либо по средней линии - на месте соединения лобных костей и костей носа, либо у внутреннего угла глаза. Внутреннее отверстие всегда одно, наружных отверстий может быть 2 и 3. Локализация наружного отверстия определяет название мозговой грыжи: носолобные, носорешетчатые и носоглазничные.
Наличие двух и более наружных отверстий дает возможность различать двусторонние и смешанные грыжи. Чаще всего встречаются двусторонние носолобные, носорешетчатые и носоглазничные мозговые грыжи.
Задние грыжи в зависимости от их отношения к затылочному бугру могут быть верхними и нижними, в зависимости от того, где расположен дефект - в затылочной кости выше или ниже затылочного бугра. Дефект кости ниже затылочного бугра нередко переходит непосредственно в большое затылочное отверстие. Чаще всего встречаются задние нижние черепно-мозговые грыжи. При этих формах в грыжевой мешок может вовлекаться измененный мозжечок. При задних верхних мозговых грыжах содержимым грыжевого мешка может оказаться затылочная доля мозга.
Базальные мозговые грыжи обычно представляют собой выбухание в полости носа или носоглотки; дефект кости находится в передней или средней черепной ямке и грыжевой мешок выпячивается в полость носа или носоглотки.
Черепно-мозговые грыжи в области сагиттального шва встречаются редко.
Клиническая картина грыжи головного мозга зависит от ее локализации и размеров. Грыжевое выпячивание, как правило, пульсирует, при пальпации в нем могут определяться как жидкость, так и плотные фиброзные включения. С течением времени грыжевое выпячивание часто увеличивается в размере, кожа над ним истончается, нередко воспаляется, может произойти разрыв истонченной стенки с развитием ликвореи, абсцесса. При носоглазничных грыжах из-за деформации и непроходимости носослезного канала часто развиваются дакриоцистит, конъюнктивит. При грыжах, выбухающих в полость носа, носовое дыхание затруднено, речь гнусавая. Грыжи головного мозга могут сопровождаться головной болью, головокружением, симптомами поражения черепных нервов, двигательными нарушениями, эпилептическими припадками, нарушением статики и походки. С возрастом нередко проявляется умственная отсталость.
Часто грыжи головного мозга сочетаются с другими пороками развития черепа и мозга:
гидроцефалией,
агенезией мозолистого тела и др.,
микроцефалией (уменьшение объема мозгового черепа).
Микроцефалия
Микроцефалия характеризуется значительным уменьшением размеров головы, гипоплазией или атрофией мозгового вещества. Вес мозга у микроцефалов может колебаться от 250 до 900г, а окружность головы уменьшается до 30-40 см. Уже в первые месяцы жизни отмечаются несоответствия в черепе: покатый лоб, выпуклые надбровные дуги и срезанный затылок. У детей раннего возраста родничок закрывается преждевременно, быстро окостеневают швы. Они прощупываются в виде выступающих валиков. Наряду с этим у больных выявляются параличи, эпилептические припадки, аномалии органов зрения (микрофтальм, коломбы, гидрофтальм, катаракта) и другие органические заболевания ц. н. с.
Типичным анатомическим признаком истинной микроцефалии являются недоразвитие больших полушарий головного мозга, непропорциональность развития отдельных частей мозга при сравнительно нормальном развитии мозгового ствола и спинного мозга.
Физическое развитие микроцефалов в большинстве случаев страдает незначительно. Слабоумие у них может быть самой различной степени - от идиотии до слабой формы олигофрении. Иногда им доступен несложный синтез и элементарная дифференциация, элементарная сообразительность. Речевые функции слабо развиты, дети произносят отдельные слоги.
На краниограммах, помимо уменьшения в размерах мозгового черепа, отмечается изменение рисунка черепных швов. Они представлены в виде тонких линий, а у детей старшего возраста могут отсутствовать. Вследствие недоразвития головного мозга кости черепа утолщаются и окостеневают швы. Внутренняя поверхность костей гладкая. В костях черепа отсутствуют признаки повышения внутричерепного давления.
Нередко встречается ложная микроцефалия. У больных при отсутствии внешних признаков микроцефалии выявляются симптомы тяжелого поражения центральной нервной системы в виде спастической гемиплегии, параплегии, тетраплегии, различных гиперкинезов и поражения черепно-мозговых нервов, которые сочетаются с выраженным умственным недоразвитием.
Этиология микроцефалии. Выделяют микроцефалию двух типов: первичную (простая, генетическая), являющуюся результатом воздействия вредного фактора на ранних стадиях внутриутробного развития, и вторичную (комбинированная, осложненная), развивающуюся в результате повреждения мозга в последние месяцы внутриутробного развития, в процессе родов и в первые месяцы жизни.
Прогноз при микроцефалии в большинстве случаев неблагоприятный. При первичной микроцефалии возможно в отдельных случаях обучение во вспомогательной школе. Дети с вторичной микроцефалией нуждаются в постоянной опеке. Хирургическому лечению микроцефалия не подлежит.
2.5 Врожденные внутричерепные аневризмы
Врожденные артериальные, венозные, артерио-венозные аневризмы представляют собой дефект развития сосудистой стенки. Гистологически определяются отсутствие, недоразвитие сосудистых стенок, дефект интимы, застойные явления в прилегающих капиллярах.
Внутричерепные аневризмы часто сочетаются с аномалиями сосудов кожи, внутренних органов, дизрафическим статусом и врожденными уродствами. Врожденные артериальные аневризмы, как правило, множественные, сопровождаются очаговой неврологической симптоматикой. Односторонний врожденный порок развития артерио-венозной системы в сочетании с пороками развития конечностей наблюдается при синдромах Клиппеля - Треноне, Паркса Вебера. Ангиоматоз мозга нередко является единственным проявлением моносимптомной формы болезни Штурге - Вебера, болезни Гиппеля - Линдау.
Внутричерепные аневризмы нередко бывают "немыми", т.е. не сопровождаются неврологическими симптомами, однако при эмоциональном возбуждении, переутомлении, при повышении внутричерепного давления они могут проявляться очаговыми симптомами. Дети жалуются на пульсирующие, давящие головные боли, быструю утомляемость, недомогание. Неврологическая симптоматика обуславливается локализацией и степенью выраженности внутричерепных аневризм. При аневризмах по ходу средней мозговой артерии часто отмечается фокальная эпилепсия в сочетании с гемипарезом, носящем переходящий характер. При разрыве аневризмы развивается клиника острого нарушения мозгового кровообращения. Лечение хирургическое.
Заключительная часть
Диагноз пороков развития головного мозга, особенно краниосхизов, обычно не вызывает затруднений. Для уточнения диагноза грыж головного мозга и некоторых пороков развития желудочковой системы необходимо дополнительное обследование, которое включает краниографию, томографию черепа, компьютерную рентгеновскую и магнитно-резонансную томографию головы, пневмоэнцефалографию, вентрикулографию, ангиографию, пункцию грыжевого выпячивания. В диагностике агенезий и пороков формирования коры большого мозга важную роль играет рентгеновская компьютерная томография. Выявление некоторых генетически обусловленных пороков развития требует специального цитогенетического и биохимического обследования. Особое значение имеют методы внутриутробной диагностики пороков развития ц. н. с.
Лечение пороков развития головного мозга оперативное. Радикальное хирургическое вмешательство осуществляют при некоторых формах врожденной гидроцефалии и в большинстве случаев мозговых грыж; его следует проводить в ранние сроки. При мозговых грыжах производят экстра - и интракраниальные операции. Задачей операций являются иссечение грыжевого мешка и пластика костного канала. При обширных дефектах кости операцию проводят в два этапа - сначала интракраниальную пластику дефекта кости черепа, а затем экстракраниальное иссечение грыжевого мешка. После операции иногда появляются рецидивы грыжи, ликворея. В случае своевременно произведенной операции большинство детей в дальнейшем развивается нормально.
Библиография
1. Бадалян Л.О. Детская неврология, Москва: Медицина, 1975.
2. Основы нейрохирургии детского возраста. Под ред. А.А. Арендта и С.И. Нерсесянц, Москва: Медицина, 1968.
3. А.П. Ромоданов, Н.М. Мосийчук Нейрохирургия, Киев: Выща школа, 1990.
4. В.Р. Пурин, Т.П. Жукова Врожденная гидроцефалия, Москва: Медицина, 1976.
5. Краткая Медицинская Энциклопедия, издание второе, Москва: Советская энциклопедия, 1989.
6. О.В. Калмин, О.А. Калмина Аннотированный перечень аномалий развития органов и частей тела человека: Учебно-методическое пособие. - Пенза: ПГУ, 2000.
7. Н.И. Федюкович Анатомия и физиология человека. Ростов-на-Дону: Феникс, 2003.
8. Внутренние болезни. В 10 книгах. Книга 10. Пер. с англ.
9. Под ред.Б. Браунвальда, К. Дж. Иссельбахера, Р.Г. Петерсдорфа и др., М.: Медицина, 1997.
Увеличивается с возрастанием срока беременности . Однако в настоящее время надежные критерии диагностики их патологии не разработаны. Важное дополнительное значение при врожденных пороках головного мозга у плода имеет сканирование в режиме ЦДK, которое позволяет оценить практически все основные сосуды головного мозга и установить сосудистый генез обнаруженных пороков, Позвоночник плода...
Симптомы и пророки развития других органов и систем.Иногда обнаружение патологии при НСГ является случайной находкой. III. Систематика методов В-сканирования головного мозгас позиций детской невропатологии и нейрохирургии В зависимости от используемых датчиков проводят линейное сканнирование или секторальное сканнирование. В зависимости от используемого ультразвукового окна различают...
При данной патологии достигает 32‰ и превосходит общий показатель в 2 раза. Эта проблема чрезвычайно актуальна, так как обуславливает серьезные последствия. У большинства женщин, перенесших гестоз, формируется хроническая патология почек, гипертоническая болезнь и эндокринные нарушения. А дети от таких матерей, как правило, имеют нарушения физического и психоэмоционального развития, при этом...
Наши представления о сущности дизонтогенеза. Кроме того, эти знания дают возможность психологу более активно и профессионально принимать участие в работе по профилактике отклонений в развитии у детей. Основные причины отклонений в психофизическом развитии и факторы их опосредующие Как известно, нарушения нервной системы могут быть вызваны как биологическими, так и социальными факторами. ...
ГЛАВА 24 ВРОЖДЕННЫЕ ДЕФЕКТЫ И АНОМАЛИИ РАЗВИТИЯ ЧЕРЕПА И ГОЛОВНОГО МОЗГА, ПОЗВОНОЧНИКА И СПИННОГО МОЗГА
24.1. ОБЩИЕ ПОЛОЖЕНИЯ
Аномалии (от греч. anomalia - отклонение, имеется в виду отклонение от нормы, от общей закономерности, неправильность) - структурные отклонения от нормы, обусловленные нарушениями пренатального развития; они представляют собой врожденные дефекты, которые проявляются уже при рождении или в раннем детском возрасте. Выраженные аномалии называют пороками развития. Пороки развития, при которых оказывается обезображенной какая-либо часть организма или все тело, иногда называют уродствами или обозначают французским словом «monstre», однако эти термины, естественно, вызывают возражения с точки зрения этики и деонтологии.
Под врожденными аномалиями подразумеваются отклонения от нормы в строении отдельных частей тела, органов и тканей. Возможны врожденные аномалии процессов метаболизма; следствием их могут быть, в частности, различные варианты олигофрении.
По этиологическому признаку различают 3 группы врожденных аномалий: а) наследственные, возникшие в результате унаследованных или спонтанных мутаций; наследственные аномалии можно разделить на геномные, хромосомные и генные; б) экзогенные, обусловленные инфекционными или токсическими тератогенными повреждениями эмбриона или плода и в) мультифакторные. К врожденным аномалиям относятся различные формы нарушения развития органов и тканей. 1. Агенезия - полное врожденное отсутствие органа. 2. Аплазия - врожденное отсутствие органа при наличии его сосудистой ножки.
3. Отсутствие или недоразвитие отдельных частей тела и органов, при этом недостаточность их развития нередко обозначается составным термином, включающим греческое слово oligos (малый) и название дефектного органа: например, олигогерия - недостаточность мозговых извилин, олигодактилия - недостаточное число пальцев. 3. Врожденная гипоплазия - недоразвитие органа, проявляющееся недостаточностью его массы или размеров. Различают простую и диспластическую формы гипоплазии. При простой форме нет качественных изменений структуры и функций органа; диспластическая же гипоплазия сказывается на функциональном состоянии органа (например, диспластическая гипоплазия глаза, или микрофтальм, сопровождаются расстройствами зрения).
4. Врожденная гипотрофия - уменьшение массы тела плода или новорожденного. 5. Врожденная гиперплазия, или гипертрофия, - относительное увеличение массы части тела или органа. 6. Макросомия (гигантизм) - увеличение тела или его части; при увеличении отдельных органов или их частей иногда при-
меняется греческий термин pachis (толстый): например, пахиакрия - утолщение фаланги пальца, пахигирия - утолщение мозговой извилины. 7. Гетерото- пия - наличие клеток, тканей или целого участка органа в другом органе или в тех частях того же органа, в которых их быть не должно, например наличие грушевидных клеток Пуркинье в зернистом слое коры мозжечка. Гетеротопия тканей характерна для некоторых опухолей, например тератомы, дермоидной кисты, холестеатомы. 8. Гетероплазия - нарушение дифференцировки ткани, также может быть основой опухолевого роста. 9. Эктопия - смещение органа, расположение его не на обычном месте. 10. Удвоение - увеличение в 2 раза количества органов или их частей; приставка «поли» (от греч. polis - много) означает увеличение их количества в неопределенное число раз, например полидактилия, полигерия. 11. Атрезия - полное отсутствие сосуда, канала или отверстия, например атрезия водопровода мозга, атрезия наружного слухового прохода. 12. Стеноз - сужение сосуда, канала или отверстия. 13. Неразделение органов, частей тела. Названия аномалий, при которых имеется неразделение конечностей или их частей, имеют приставку «sym» или «syn» (вместе), например симподия - неразделение ног, синдактилия - неразделение пальцев. Возможно и неразделение двух симметрично или асимметрично развитых однояйцевых близнецов. Неразделившуюся двойню («сиамские близнецы») называют пагами, добавляя к этому слову латинское название частей тела, которыми они соединены, например при сращении головами - краниопаги (см. рис. 24.3), грудной клеткой - торакопаги и т.п. 14. Персистирование - сохранение структур, в норме исчезающих к определенному периоду эмбрионального развития. Персистирование эмбриональной ткани может стать причиной развития опухолей, возникающих вследствие дизэмбриогенеза (по теории Конгейма), например краниофарингиома. 15. Дизрафия - незаращение эмбриональной срединной щели - незаращение верхней губы, нёба, дужек позвонков и т.д. 16. Инверсия - обратное (зеркальное) расположение органов.
Пренатальное, в частности эмбриональное, развитие нервной системы - сложнейший процесс, который может быть нарушен под влиянием разных причин, в том числе унаследованных особенностей генофонда и эндогенных или экзогенных влияний, прежде всего внутриутробных травм, инфекции и интоксикации. Характер возникающих при этом аномалий во многом зависит от фазы развития нервной системы: стадии формирования нервной трубки (первые 3,5-4 нед), формирования мозговых пузырей (4-5 нед), коры больших полушарий (6- 8 нед) и т.д. Вследствие этих причин могут возникать разнообразные дефекты развития головного и спинного мозга, черепа и позвоночника. Эти пороки могут встречаться изолированно или в различных сочетаниях.
Вторичные нарушения развития и деформации черепа и мозга во внутриутробном периоде, во время родов или в раннем детстве, а также в более позднем возрасте могут быть следствием травматических повреждений, инфекционных заболеваний, а иногда и неуточненных обстоятельств. Вторичные деформации тканей головы и мозга могут быть обусловлены преждевременным сращением черепных костей, гидроцефалией, рахитом, болезнью Педжета, мраморной болезнью и пр.
На долю нарушений развития ЦНС приходится более 30% всех аномалий, обнаруживаемых у детей (Huidi C., Dixian J., 1980). Частота врожденных аномалий развития ЦНС варьирует, средний ее показатель 2,16 на 1000 родившихся.
24.2. КРАНИОСИНОСТОЗ, КРАНИОСТЕНОЗ
Одной из причин аномалий черепа является преждевременное и иногда неравномерное окостенение черепных швов - краниосиностоз (от греч. kranion - череп и sinostosis - сращение). В норме у новорожденных детей все кости свода черепа не сращены, передний и задний родничок открыты. Задний родничок закрывается к концу 2-го месяца, передний - в течение 2-го года жизни. К концу 6-го месяца жизни кости свода черепа соединяются между собой плотной фиброзной мембраной. К концу 1-го года жизни размер головы ребенка составляет 90%, а к 6 годам он достигает 95% от размера головы взрослого че- ловека. Смыкание швов путем соединения зубчатых краев костей начинается к концу 1-го года жизни и полностью заканчивается к 12-14-летнему возрасту.
Преждевременное и неравномерное зарастание родничков и черепных швов у детей ведет к развитию краниостеноза (от греч. kranion - череп и stenosis - сужение) и, следовательно, к недостаточности объема полости мозгового черепа, препятствующей нормальному развитию мозга и ведущей к созданию условий для ликвородинамических нарушений. Частота краниостеноза 1 на 1000 новорожденных. При краниостенозе обычно повышено внутричерепное давление, в связи с этим характерна гипертензионная головная боль, возможны развитие застойных дисков зрительных нервов с последующей их вторичной атрофией и нарушением зрения, отставание в умственном развитии (подробнее о внутричерепной гипертензии см. в главе 20).
Различают первичный (идиопатический) и вторичный краниосиностозы. Раз- витие вторичного краниосиностоза может быть обусловлено различными причинами. К ним могут быть отнесены витамин D-дефицитный рахит, гипофосфатемия, передозировка тиреоидного гормона в случаях лечения врожденной гипотиреоидной олигофрении (кретинизма).
Зарастание швов черепа не только преждевременное, но и неравномерное обычно ведет к деформации черепа. В процессе контроля за развитием формы мозгового черепа учитывается так называемый черепной индекс (ЧИ) - соотношение поперечного размера черепа к продольному его размеру, умноженное на 100. При нормальном (среднем) соотношении поперечного и продольного размеров головы (при мезоцефалии) черепной индекс у мужчин составляет
76-80,9, у женщин - 77-81,9.
При преждевременном зарастании сагиттального шва (сагиттальном синостозе) развивается долихоцефалия, при которой череп увеличивается в переднезаднем направлении и оказывается уменьшенным в поперечном размере. В таких случаях голова оказывается узкой и удлиненной. ЧИ при этом меньше 75.
Вариант долихоцефалии, обусловленной преждевременным заращением сагиттального шва (рис. 24.1), при котором возникает ограничение роста черепа в поперечном направлении и оказывается избыточным рост его в длину, могут быть скафоцефалия (от греч. skaphe - лодка), цимбоцефалия (ладьевидная голова, килеголовость), при которой формируется длинная узкая голова с выступающими лбом и затылком, напоминающая перевернутую вверх килем лодку. Седловидным называется удлиненный в продольном направлении череп с вдавлением в теменной области.
Вариантом деформации черепа, при которой череп имеет увеличенный поперечный размер в связи с преждевременным заращением коронарных (венечных) швов (коронарный, или венечный, синостоз), является брахицефалия (от греч. brachis - короткий и kephale - голова), голова при этом широкая и
Рис. 24.1. Скафокрания у ребенка 5 лет.
укороченная, черепной индекс свыше 81. При брахицефалии в связи с двусторонним коронарным синостозом лицо уплощено, нередко проявляется экзофтальм.
При преждевременном заращении венечного шва с одной стороны развивается плагиоцефалия, или косоголовость (от греч. plagios - косой и kephale - голова). В таких случаях череп асимметричен, лобная кость на стороне синостоза уплощена, на этой же стороне возможны экзофтальм и увеличение средней и задней черепных ямок.
Если возникает преждевременное сочетанное заращение коронарного и сагиттального черепных швов, рост черепа происходит в основном в стороны переднего родничка и основания, что приводит к увеличению высоты головы при ограничении ее роста в продольном и поперечном на- правлениях. В результате формируется высокий череп конической формы, несколько уплощенный в переднезаднем направлении (акрокрания), его часто называют башенным черепом (рис. 24.2). Вариант башенного черепа - оксицефалия, или остроконечная голова (от греч. oxys - острый, kephale - голова), при которой раннее зарастание черепных швов ведет к формированию высокого, суживающегося кверху черепа со скошенным назад лбом.
Вариант деформации черепа, характеризующийся узкой лобной и широкой затылочной костями, формируется в связи с преждевременным зарастанием
лобного шва. Лобные кости при этом срастаются под углом (в норме зарастание лобного шва происходит только к концу 2-го года жизни) и на месте лобного шва формируется «гребень». Если в таких случаях компенсаторно увеличиваются задние отделы черепа и углубляется его основание, возникает тригонокрания, или треугольный череп (от греч. trigonon - треугольник, kephale - голова).
Изолированный синостоз ламбдовидного шва встречается крайне редко и сопровождается уплощением затылка и компенсаторным расширением передней части черепа с увеличением переднего родничка. Нередко он сочетается с преждевременным закрытием сагиттального шва.
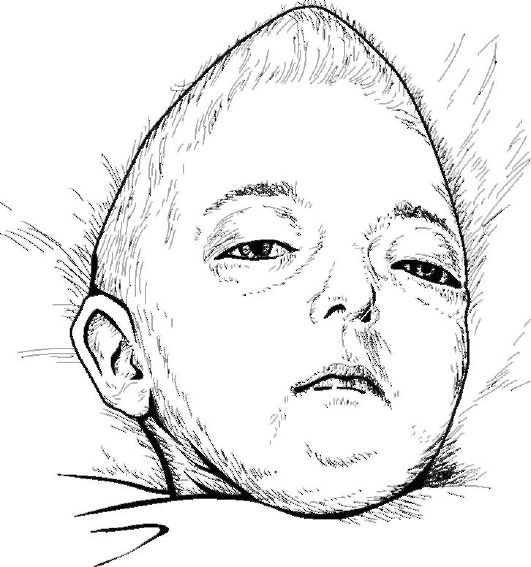
Рис. 24.2. Башенный череп у ребенка 3 лет.
Примером сочетания генетически обусловленного краниостеноза с другими патологическими проявлениями может быть симптомокомплекс Терсиля (описал в 1942 г. французский врач Thersil M.): башенный череп, экзофтальм, нистагм, олигофрения, эпилепсия, атрофия зрительных нервов. На краниограммах обычно имеются проявления внутричерепной гипертензии, в частности выраженные пальцевые вдавления.
При вторичном краниостенозе на раннем этапе его развития может быть эффективно консервативное лечение основного заболевания. При первичном краниостенозе, а также при вторичном краниостенозе в случае уже развившейся значительной внутричерепной гипертензии показана декомпрессивная операция: формирование краниоэктомических проходов шириной до 1 см по линии шовных окостенений. Своевременное хирургическое лечение при кра- ниостенозе может обеспечить в дальнейшем нормальное развитие мозга.
24.3. ГИПЕРТЕЛОРИЗМ И ГИПОТЕЛОРИЗМ
Одним из вариантов аномалии черепа является гипертелоризм (от греч. tele - далеко, horismos - разграничение, разделение), представляющий собой следствие чрезмерного развития малых крыльев основной кости. Значительно увеличено расстояние между внутренними краями орбит, широкая переносица, плоская спинка носа, широко расставленные глаза. Может сочетаться с микроофтальмией, эпикантусом, двусторонним сходящимся косоглазием, другими аномалиями, отставанием в психическом развитии.
Семейные формы гипертелоризма наследуются по аутосомно-доминантному типу. Гипертелоризм может быть одним из признаков наследственных болезней, имеющих различный тип передачи (синдромы Крузона, Грега, «кошачьего крика» и пр.).
При гипертелоризме индекс межорбитально-окружностный (ИМО) более 6,8. ИМО равен результату деления расстояния (в сантиметрах) между внутренними углами глазных щелей на окружность головы, умноженному на 100.
Гипотелоризмом принято называть уменьшение расстояния между внутрен- ними краями глазниц; при этом возможно недоразвитие носа, лицо похоже на морду обезьяны, ИМО меньше 3,8. Гипертелоризм может быть одним из при- знаков некоторых наследственных заболеваний, например синдрома Патау.
24.4. МАКРОКРАНИЯ, МИКРОКРАНИЯ, КРАНИОТАБЕС, КРАНИОСКЛЕРОЗ
Увеличение размеров черепа (макрокрания) может быть не только врожденным, но и приобретенным, например при рахите, несовершенстве остеогенеза, черепно-ключичном дизостозе.
У новорожденных возможна асимметричная макрокрания и в связи с субдуральной гематомой, гигромой, арахноидальной кистой в области свода черепа. Асимметрия черепа при гемиатрофии мозга вследствие перенесенного в раннем детстве травматического или воспалительного его поражения, сопровождающаяся уплощением, иногда утолщением костей свода черепа, известна как
симптом Копылова (описал отечественный нейрорентгенолог Копылов М.Б., род. в 1887 г.). Надо иметь в виду, что асимметрия черепа при рождении может быть и следствием подкожной или подапоневротической гематомы.
При рахите, обычно при остром его течении, иногда возникает краниотабес - размягчение, истончение плоских костей черепа в области переднего и заднего родничков, над сосцевидными отростками и по ходу черепных швов. Возможно также развитие гиперостоза черепа (краниосклероз) - медленно прогрессирующее утолщение и неравномерное увеличение размеров костей черепа, чаще лицевого; наблюдается, например, при паратиреоидной остео- дистрофии, нейрофиброматозе, эозинофильной аденоме гипофиза (сомато- тропиноме), при опухолях костей черепа.
24.5. КРАНИОПАГИЯ
Краниопагия относится к числу наиболее редких и опасных врожденных уродств; она представляет собой срастание двух однояйцевых близнецов головами (рис. 24.3).
Разделение краниопагов относится к сложнейшим нейрохирургическим вмешательствам, включающим разделение мозга обоих младенцев, кровоснабжающих их мозг сосудов, твердой мозговой оболочки, кожных покровов и осуществление сложных реконструктивных операций для замещения неизбежных при разделении близнецов дефектов костей черепа и мягких тканей головы. В литературе описано около 30 операций по разделению краниопагов, эти операции, к сожалению, чаще заканчиваются гибелью одного или обоих близнецов. Опыт успешной операции по разделению краниопагов принадлежит Институту нейрохирургии им. Н.Н. Бурденко РАМН.

Рис. 24.3. Сиамские близнецы, сросшиеся головами, - краниопаги.
24.6. ПЛАТИБАЗИЯ
Аномалия развития черепа, проявляющаяся уплощением его основания, - платибазия (от греч. platys - плоский и basis - основание). Она может быть и следствием проявившейся в детском возрасте длительной внутричерепной гипертензии. При платибазии особенно уплощена задняя черепная ямка, обычно сильно увеличено расстояние между спинкой турецкого седла и большим затылочным отверстием; угол, образованный скатом черепа (блюменбахов скат) и передней частью основания черепа (фронтальное основание, плоскость передней черепной ямки), больше 105?; передний край большого затылочного отверстия и передняя дуга атланта несколько приподняты (рис. 24.4б). Платибазия иногда протекает бессимптомно, но может сопровождаться повышением внутричерепного давления. Врожденная платибазия наблюдается при болезни Дауна, мукополисахаридозах, может сочетаться с аномалией Арнольда-Киари, ахондропатией. Приобретенная платибазия возможна при болезни Педжета, остеомаляции, фиброзной дисплазии, гипотиреозе, она может сопровождаться базилярной импрессией.
24.7. БАЗИЛЯРНАЯ ИМПРЕССИЯ
Базилярная импрессия (базилярная инвагинация, базилярное вдавление) обычно возникает на фоне врожденной платибазии и представляет собой углубление переднего отдела основания затылочной кости (краев большого затылочного отверстия, затылочных мыщелков) в сторону субтенториального пространства. На краниограммах при этом можно отметить увеличение угла между скатом и верхней пластинкой основной кости (более 130?, рис 24.4в), а также смещение верхних шейных позвонков, прежде всего зуба II шейного (осевого) позвонка выше линии Чемберлена (условная линия, соединяющая задний край твердого нёба с задним краем затылочного отверстия, определяемая на профильной краниограмме) и линии де ля Пти (условная линия между верхушками сосцевидных отростков, определяемая на фасной краниограмме). Обычно у таких больных короткая шея, ограничение ее подвижности, низко расположенная граница роста волос на шее. В первое-второе десятилетие жизни возможны клинические проявления нарушения функций структур, расположенных в задней черепной ямке, и верхних шейных сегментов спинного мозга (спастический тетрапарез, элементы бульбарного синдрома, нистагм при повороте взора вниз - нистагм, «бьющий вниз», и пр.), а также нарушения ликвородинамики, проявляющиеся гидроцефалией (см. синдром Арнольда-Киари-Соловце- ва, глава 11).
24.8. ПОДВЫВИХ В АТЛАНТООСЕВОМ СУСТАВЕ
Фактором риска является нестабильность в атлантоосевом суставе. В таких случаях даже легкая травма может привести к его подвывиху и глубокому неврологическому дефекту, обусловленному компрессией спинномозговых корешков C I -C II и соответствующих нервов, а также позвоночных артерий и орального отдела спинного мозга. В случае возможного при этом вклинения
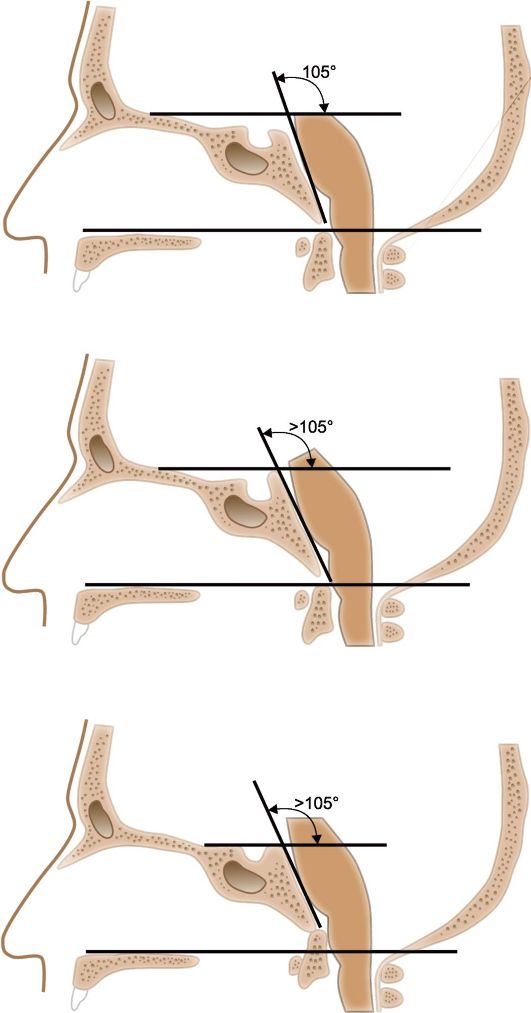
Рис. 24.4. Определение платибазии и базилярной импрессии.
а - в норме: твердое нёбо, верхушка зуба осевого (II шейного) позвонка и край большого затылочного отверстия расположены на одной линии или верхушка зуба осевого позвонка находится ниже этой линии, а угол, образованный основанием передней черепной ямки и скатом, равен приблизительно 105 градусам; б - платибазия: угол наклона ската по отношению к основанию передней черепной ямки более 105 градусов; в - базиляр- ная импрессия: верхушка зуба осевого позвонка выше линии, проходящей через твердое нёбо и край затылочного отверстия; угол наклона ската больше 105 градусов.
зубовидного отростка II шейного (осевого) позвонка в большое затылочное отверстие обычно наступает смерть от остановки дыхания. Предрасположенность к подвывиху атлантоосевого сустава имеется при синдроме Дауна, ревматоидном артрите, мукополисахаридозе.
24.9. АКРОЦЕФАЛОСИНДАКТИЛИИ
Многовариантную группу врожденных аномалий составляют различные формы сочетаний башенного черепа (акрокрания, акроцефалия) с различными вариантами аномалии пальцев (акроцефалосиндактилии, акроцефалополисиндактилии).
24.10. СИНДРОМ ГРУБЕРА
Среди других наследственных заболеваний, сопровождающихся выраженной костной патологией, в частности изменениями черепа, можно отметить синдром Грубера, проявляющийся микроцефалией, уплощением глазниц, экзофтальмом, пороками развития лицевого скелета, нередко расщеплением дужек позвонков, оболочечными и оболочечно-мозговыми грыжами на спинальном уровне. Этот синдром наследуется по аутосомно-рецессивному типу. Описал его в 1933 г. H. Gruber.
24.11. ОКОНЧАТЫЕ ДЕФЕКТЫ ЧЕРЕПА
На краниограммах иногда удается обнаружить небольшие врожденные окончатые дефекты черепа, локализующиеся в сагиттальной плоскости или парасагиттально, преимущественно в теменной области. Окончатые дефекты черепа иногда сочетаются с проявлениями дизрафии, в частности, дизрафии дужек позвонков.
24.12. ДИЗОСТОЗЫ ЧЕРЕПА
Деформации черепа могут быть проявлением различных вариантов дизостоза.
Черепно-лицевой дизостоз Крузона, или «попугайная» болезнь, - краниостеноз, обусловленный сочетанием недоразвития костей черепа и преждевременным зарастанием черепных швов. Проявляется изменением формы мозгового и лицевого черепа, при этом характерны гипертелоризм, экзофтальм, страбизм, своеобразная крючковатая форма носа, напоминающего клюв орла или по- пугая. Возможны недоразвитие нижней челюсти, нарушение прикуса: нижние зубы впереди верхних (прогнатия), снижение слуха, пирамидная и мозжечковая недостаточность, реже - другие очаговые неврологические симптомы. Могут быть различные аномалии костей туловища и конечностей. На глазном дне нередко отмечаются признаки застоя, который может смениться вторичной атрофией дисков зрительных нервов, сопровождающейся нарушением зрения.
Наследуется по аутосомно-доминантному типу. Описал в 1912 г. французский врач O. Crouzon (1874-1938).
Черепно-лицевой дизостоз Франческетти-Цвалена характеризуется грубыми нарушениями строения мозгового и лицевого отделов черепа («рыбье лицо»). Лицо вытянуто, разрез глаз антимонголоидный, верхняя и нижняя челюсти с обеих сторон недоразвиты, отмечаются гипоплазия структур пирамид височных костей, деформации ушных раковин, выраженное снижение слуха, иногда вплоть до глухоты. Нередко сочетается с другими пороками развития. Насле- дуется по аутосомно-доминантному типу.
Черепно-ключично-тазовый дизостоз Шенте-Мари-Сентона - семейное за- болевание, характеризующееся запаздывающим зарастанием черепных швов и родничков, брахицефалией, выраженным гипертелоризмом, гиперостозом дна средней черепной ямки, отсутствием пневматизации пирамид височных костей, недоразвитием верхних челюстей и гайморовых пазух, запоздалым развитием и дистрофией постоянных зубов, частичным или полным недоразвитием ключиц (вследствие чего плечевые суставы можно сближать на груди до их соприкосновения), сколиозом, глубоким поясничным лордозом, иногда расщеплением дужек позвонков, спинномозговыми грыжами. Возможны проявления сдавливания плечевых сплетений. Грудная клетка конической формы, таз узкий, позднее окостенение лобковых костей, брахидактилия, брахимезофалангия, иногда прогрессирующее снижение слуха. При рентгенографии выявляют склероз костной ткани, деформации костей, множественные шпоровидные костные утолщения. Наследуется по аутосомно-доминантному типу. Возможны и спорадические случаи. Описали в 1898 г. J. Shentaner, Р. Marie и R. Sainton.
24.13. ПАТОЛОГИЯ ЧЕРЕПА ПРИ СИСТЕМНЫХ
ЗАБОЛЕВАНИЯХ КОСТЕЙ
Некоторые неврологические расстройства сопряжены с системными заболеваниями костей, с которыми в связи с этим должен быть знаком врач-невропатолог, поэтому ниже приводится краткая информация о такого рода костной патологии.
Для фиброзной остеодисплазии, или болезни Брайцева-Лихтенштайна, характерно нарушение костеобразующей функции мезенхимы, проявляющееся в одной или нескольких костях, что ведет к их деформации и образованию в них очагов разрежения, обычно отграниченных от здоровой ткани кости склеротической каймой. Объем пораженной кости при этом может быть увеличен. Чаще поражаются трубчатые кости, но характерные изменения могут отмечаться и в костях черепа. В таких случаях возможны облитерация прида- точных полостей носа, деформация глазниц, сужение отверстий в основании мозгового черепа и в лицевом черепе, ведущее к нарушению функции проходящих через них нервов и сосудов. Заболевание, возможно, наследственное, проявляется с детских лет. Описал в 1927 г. отечественный хирург В.Р. Брайцев (1878-1964), несколько позже - американский патологоанатом L. Liechtenstein (1906-1977).
Деформирующая остеодистрофия (болезнь Педжета) чаще проявляется у мужчин в возрасте 40-60 лет, характеризуется постепенно прогрессирующим
утолщением коркового слоя костей с развитием гиперостозов, деформацией, искривлением костей, беспорядочностью их структуры, образованием в них кист; поражаются кости мозгового черепа, позвоночника и длинных трубчатых костей. Размеры мозгового черепа увеличиваются, наружная пластинка костей свода черепа местами утолщена, гиперостозы чередуются с участками беспорядочного разрежения кости. В связи с деформацией костных от- верстий и каналов основания черепа и межпозвонковых отверстий нарушается функция черепных и спинномозговых нервов, возможны расстройства кровообращения. Деформация глазниц обусловливает экзофтальм. Нередко отмечаются признаки внутричерепной гипертензии. Позвонки сплющены; в трубчатых костях сужены костномозговые каналы, возможны патологические переломы костей, при этом линия перелома четкая, ровная, как при переломе очищенного банана («банановый перелом»); усилены физиологические изгибы позвоночника. Процесс может быть относительно ограниченным или распространенным. Содержание кальция и фосфора в крови нормально или слегка увеличено, активность щелочной фосфатазы повышена. Предполагается доминантный тип наследования с различной экспрессивностью. Описал болезнь в 1877 г. английский хирург J. Paget (1814-1899).
Мраморная болезнь (болезнь Альберс-Шенберга) - семейный генерализованный остеосклероз, протекающий с лейкемической реакцией крови у детей, с анемией и лейкопенией у взрослых, нередко с атрофией зрительных нервов и глухотой. Характерны деформация мозгового и лицевого черепа, заращение придаточных полостей носа плотной бесструктурной костной тканью. Ввиду постепенного сужения отверстий в черепе и межпозвонковых отверстий могут возникать полиморфные проявления поражения периферической нервной системы как на черепном, так и на позвоночном уровнях. В позвонках костные балки губчатого вещества утолщены и уплотнены. В трубчатых костях отмечается сужение, а затем и исчезновение костномозговых полостей, эпифизы булавовидно утолщены и поперечно исчерчены, имеется склонность к патологическим переломам. Наследуется по аутосомно-рецессивному типу и тогда, проявляясь в фенотипе в первые годы жизни, быстро приводит к смерти, или же - по аутосомно-доминантному типу, проявляясь в 20-40-летнем возрасте. Описал болезнь в 1907 г. H.E. Abers-Schonberg.
Синдром Олбрайта представляет собой множественную фиброзную дисплазию костей, сопровождающуюся болями и спонтанными переломами; при этом возможны повреждения верхней стенки глазницы. В таких случаях отмечается односторонний экзофтальм, на той же стороне - атрофия зрительного нерва, офтальмопарез. Обычны головная боль, расстройство слуха, судороги, олигофрения, гипертиреоз, зоны кожной гиперпигментации. Проявляется в детском возрасте. У девочек при этом возможно преждевременное половое созревание (менструации начинаются в 5-8 лет). Этиология неизвестна. Описали синдром в 1937 г. американский эндокринолог F. Albright (род. в 1900 г.) и соавт.
Энцефалоофтальмическая семейная дисплазия Краузе-Ризе - эктомезодермальная дисплазия, проявляющаяся сразу после рождения главным образом неврологическими и офтальмологическими симптомами. Характерны долихоцефалия, иногда гидроцефалия, затылочная или пояснично-крестцовая грыжа, мозжечковая атаксия, абсансы, олигофрения, раздражительность, а также птоз верхних век, страбизм, миопия, отслойка сетчатки, катаракта. Возможны расщепление верхней губы, твердого нёба, врожденные пороки сердца и другие дефекты развития. Наследуется по аутосомно-доминантному типу. Описали
эту форму патологии в 1946 г. австрийский врач A.C. Krause и в 1958 г. американский офтальмолог A.B. Reese.
Краниометафизарная дисплазия - диффузное разрастание костной ткани черепа и метафизов трубчатых костей. Характерны большая голова, гипертелоризм, седловидный нос, широко расставленные зубы. Сужение отверстий основания черепа может обусловить поражение черепных нервов и сосудистые расстройства. Ноги обычно непропорционально длинные, их суставные зоны утолщены. Течение заболевания медленно прогрессирующее. Наследуется по аутосомно-рецессивному типу. Описал этот патологический процесс в 1957 г. O. Lehman.
Синдром Дзержинского - семейная гиперпластическая периостальная дис- трофия, проявляющаяся комбинацией пороков развития, при этом характерны различные варианты краниосиностоза, базилярная импрессия. Кости мозгового черепа и лица утолщены, уплотнены, нос резко выступающий, утолщены ключицы, грудина, иногда наблюдается воронкообразная грудь, пальцы короткие, их фаланги утолщены. Синдром, вероятно, наследственный. Описал заболевание в 1913 г. польский врач В.Э. Дзержинский.
При хроническом ксантоматозе, или болезни Хенда-Шюллера-Христиана, характерна триада Крисчена: дефекты в костях черепа, экзофтальм и несахарный диабет. В черепе, а также в позвонках и трубчатых костях, развивается ретикулогистиоцитарная пролиферация с образованием гранулем и последующим рассасыванием костной ткани. Над очагами костной деструкции сначала возникают плотные болезненные выбухания, затем в той же зоне образуются кратерообразные углубления. Разрушение основания черепа и глазниц может сопровождаться опущением глазных яблок. Сдавление гранулематозными массами мозга и черепных нервов ведет к развитию разнообразной неврологической симптоматики. На краниограмме кости черепа изменены по типу «географической карты» (в связи с очагами остеопороза с неровными контурами). В основе лежит генетически обусловленное нарушение липоидного обмена с образованием опухолевидных скоплений жиролипоидных масс в различных органах и тканях. В крови при этом выявляются признаки гипохромной анемии, повышено содержание холестерина и липопротеинов. Болезнь проявляется в детском возрасте (до 10 лет), чаще у мальчиков. Наследуется по аутосомно- рецессивному типу. Описал болезнь в 1933 г. американский педиатр A. Hand (род. в 1868 г.), затем - американский врач H.A. Christian (1876-1951) и авс- трийский рентгенолог A. Schuller (род. в 1874 г.).
Синдром Ван-Бюхема - наследственный генерализованный гиперостоз, проявляющийся после наступления половой зрелости умеренными признаками акромегалии. С 3-го десятилетия жизни появляются экзофтальм, ухудшение слуха, периферические парезы лицевых нервов. На рентгенограммах отмечаются проявления генерализованного гиперостоза, в крови - повышение уровня щелочных фосфатаз, нормальное содержание кальция и фосфора. Описал синдром в 1952 г. голландский терапевт F. van Buchem.
Гипопластическая хондродистрофия представляет собой врожденную болезнь, характеризующуюся нарушением энхондрального остеогенеза. Характерны большой мозговой череп с выступающим затылком, седловидный нос, прогнатизм, низкий рост (у взрослых до 130 см) в основном за счет укорочения конечностей (микромиелический нанизм), короткие кисти, выражен поясничный лордоз. Возможны корешковые боли, нижний парапарез, обструктивные апноэ во сне. При рождении длина тела 46-48 см, отмечается значительное отставание моторного развития, возможно умеренное отставание умственно-
го развития. На рентгенограммах выявляются диспропорция мозгового и лицевого черепа, уплощение основания черепа, укорочение трубчатых костей, утолщения подвздошных костей, крылья которых развернуты, сужение позвоночного канала. Тип наследования аутосомно-доминантный, в 80% случаев заболевание обусловлено новыми мутациями.
Дизрафический синдром, или синдром Бремера, представляет собой комп- лекс дефектов эмбриогенеза, расположенных преимущественно вдоль средней линии: высокое нёбо, расщепление нёба и верхней губы («волчья пасть» и «заячья губа»), неравномерный рост и неправильное расположение зубов, деформации черепа, грудной клетки, кранио-вертебральные аномалии, проявления сирингомиелии, деформации позвоночника, расщепление дужек позвонков (spina bifida), спинномозговые и черепные оболочечные и оболочечно-мозговые грыжи, добавочные и несимметричные грудные железы, ночное недержание мочи.
24.14. ЧЕРЕПНО-МОЗГОВЫЕ ГРЫЖИ
Врожденным пороком развития являются черепно-мозговые грыжи, которые встречаются с частотой 1:4000-5000 новорожденных. Эта форма порока развития формируется на 4-м месяце внутриутробного развития. Она представляет собой грыжевое выпячивание в области костного дефекта, который может быть различным по своему размеру и форме. Локализуются грыжи обычно в местах соединения костей черепа: между лобными костями, у корня носа, около внутреннего угла глаза (передние грыжи), в области соединения теменных костей и затылочной кости (задние грыжи). Чаще других встречаются передние черепномозговые грыжи (рис. 24.5). По локализации наружного отверстия грыжевого канала они дифференцируются на носолобные, носорешетчатые и носоглазнич-
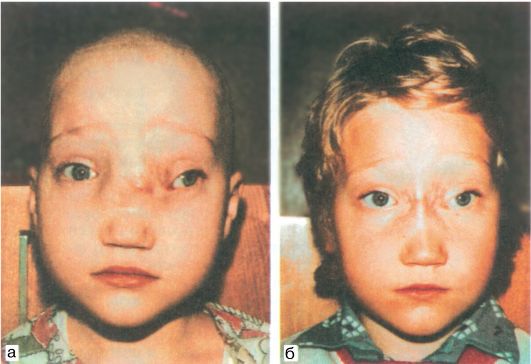
Рис. 24.5. Ребенок с назоорбитальной грыжей и гипертелоризмом до (а) и после (б) операции.
Рис. 24.6. Ребенок с грыжей в затылочной области.
ные. Задние черепно-мозговые грыжи (рис. 24.6) разделяются на верхние и нижние в зависимости от того, где расположен дефект в затылочной области: выше или ниже затылочного бугра. Кроме названных вариантов черепномозговых грыж, иногда выявляются так называемые базальные грыжи, при которых имеется дефект костей основания черепа на дне передней или средней черепных ямок, и грыжевой мешок выпячивается в полость носа или носоглотки. Редко встречаются черепномозговые грыжи в области сагиттального шва.
Основными формами черепно-мозговых грыж являются: 1) менингоцеле, при которой грыжевой мешок представлен кожей и измененными мягкой и паутинными оболочками, твердая мозговая оболочка обычно не принимает участия в образовании грыжевого выпячивания, а фиксируется к краям дефекта кости; содержимым грыжевого мешка при этом является ЦСЖ; 2) ме- нингоэнцефалоцеле - грыжевой мешок составляют те же ткани, а содержимое его, кроме ЦСЖ, составляет и ткань мозга; 3) менингоэнцефалоцистоцеле - грыжевое выпячивание, в которое, кроме тех же тканей, вовлекается и часть расширенного желудочка мозга. Из перечисленных трех форм черепно-мозговых грыж чаще встречается менингоэнцефалоцеле, нередко именуемое как энцефалоцеле. При гистологическом изучении грыжевого мешка и его содержимого выявляются утолщение и уплотнение (фиброз) мягкой и паутинной оболочек, резкая атрофия и перерождение оказавшейся в грыжевом мешке мозговой ткани.
Поверхность грыжевого выпячивания может быть покрыта неизмененной кожей или истонченной, рубцово-измененной кожей, имеющей синеватую окраску. Иногда уже при рождении ребенка в центре грыжи имеется ликворный свищ. Нередко в первые годы жизни ребенка размеры грыжевого выпячивания значительно увеличиваются, при этом его кожные покровы истончаются и изъязвляются. Возможен и разрыв грыжевого мешка с массивной ликвореей, опасной для жизни. К тому же изъязвления на поверхности грыжевого мешка и ликворные свищи зачатую инфицируются, что может обусловить развитие гнойного менингоэнцефалита. Грыжевое выпячивание бывает на ножке (заужено в основании) или же имеет широкое основание. В последнем случае оно нередко пульсирует, а при натуживании ребенка - напрягается. При пальпации грыжевое выпячивание может быть различной плотности, эластичным, флюктуирующим.
Передние черепно-мозговые грыжи вызывают обезображивание лица, деформацию глазниц, носа, при этом нередко отмечаются уплощенная широкая переносица, неправильное расположение глазных яблок, нарушение бинокулярного зрения. При назоорбитальных грыжах, как правило, выявляются деформация и непрохо-
димость слезно-носового канала, часто развиваются конъюнктивит, дакриоцистит. Базальные черепно-мозговые грыжи, располагающиеся в полости носа или носоглотки, по внешнему виду напоминают полипы. Если грыжевой мешок находится в одной половине носа, возникает искривление носовой пере- городки; при этом дыхание затруднено, речь невнятная с носовым оттенком.
Очень крупные менингоэнцефалоцеле (есть описание передней черепно-мозговой грыжи диаметром 40 см) обычно сопровождаются выраженной мозговой патологией, и новорожденные в таких случаях оказываются нежизнеспособными. Судьба остальных больных, как правило, зависит от размеров и содержимого грыжевого выпячивания, а также возможности оперативного лечения этого порока развития. Дети нередко испытывают головную боль, головокружение. Очаговая мозговая симптоматика может отсутствовать или быть умеренно выраженной, однако возможны и очаговые неврологические симптомы, в частности центральные парезы, гиперкинезы, расстройства координации движений и пр., признаки недостаточности функций черепных нервов (I, II, VI, VII, VIII, XII). Возможны эпилептические пароксизмы, отставание умственного развития.
Черепно-мозговые грыжи могут сочетаться с другими врожденными аномалиями: микроцефалией, краниостенозом, гидроцефалией, микрофтальмией, эпикантусом, врожденным птозом верхнего века, аномалией развития сетчатой оболочки глаза и зрительных нервов, колобомами (дефекты тканей глазного яблока), врожденным гидрофтальмом, краниоспинальными аномалиями, расщеплением дужек позвонков.
Лечение мозговых грыж. Показаниями к безотлагательной операции у но- ворожденного являются ликворея из грыжевого мешка или быстрое увеличение размеров грыжи с истончением ее покровов и опасностью разрыва. При отсутствии срочных показаний к операции ребенок должен находиться под наблюдением педиатров, невропатологов, нейрохирургов, которые обычно совместно решают вопрос о возможности оказания больному нейрохирургической помощи и определяют наиболее благоприятные сроки операции. Надо иметь в виду, что оперативное лечение черепно-мозговой грыжи может быть эффективным и нередко приводит к благоприятному результату (рис. 24.5).
Противопоказаниями к операции являются воспалительные процессы в оболочках и в головном мозге, выраженные неврологические и психические расстройства (имбецильность, идиотия), проявления гидроцефалии, тяжелые сопутствующие уродства.
Хирургическое лечение заключается в выделении и иссечении грыжевого мешка с сохранением при этом его содержимого. Важными этапами операции являются герметичное зашивание твердой мозговой оболочки и тщательная пластика костного дефекта.
При сочетании носоглазничной грыжи и гипертелоризма выполняется сложная реконструктивная операция, включающая пластику костного дефекта и сближение глазниц. Затылочные мозговые грыжи могут содержать венозные синусы твердой мозговой оболочки, что необходимо иметь в виду при хирургическом вмешательстве.
24.15. ПОРОКИ РАЗВИТИЯ ГОЛОВНОГО МОЗГА
Пороки развития могут проявляться в различных сочетаниях. Так, например, при синдроме Дуранда-Дзунина признаки дизрафии сочетаются с гидроцефалией, сопровождающейся увеличением мозгового черепа, агенезией
прозрачной перегородки, расщеплением дужек позвонков, искривлением стоп и двусторонней гипоплазией почек, ведущей к нарушению водного обмена. Синдром имеет семейный, по-видимому, наследственный характер. Описали его в 1955 г. итальянские педиатры S. Durand и F. Zunin.
В особую группу аномалий развития могут быть выделены выраженные
вторичные врожденные пороки развития черепа и мозга, возникшие в различные периоды онтогенеза. Причины таких аномалий многообразны: заболевания ма- тери в период беременности, облучение, травматические повреждения плода, воздействие на плод разнообразных токсических факторов, в частности алкоголя и многочисленных лекарственных препаратов, оказывающих тератогенное действие. Пороки развития ЦНС являются следствием одного или нескольких основных патологических процессов, нарушающих развитие мозга: образование нервной трубки, разделение ее краниального отдела на парные образования, миграция и дифференциация клеточных элементов нервной ткани. Они могут проявляться на трех уровнях: клеточном, тканевом и органном.
Ниже приводится описание некоторых дефектов развития головного мозга и черепа, возникающих в процессе онтогенеза (вследствие дизэмбриогенеза).
Анэнцефалия - отсутствие большого мозга, костей свода черепа и покрывающих его мягких тканей. На месте мозгового вещества обычно располагается соединительная ткань, богатая кровеносными сосудами, с кистозными полостями, выстланными медуллярным эпителием, глиальная ткань, единичные не- рвные клетки, остатки сосудистых сплетений.
Эксэнцефалия - отсутствие костей свода черепа (акрания) и мягких покровов головы, в результате чего большие полушария располагаются открыто на основании черепа в виде отдельных узлов, покрытых мягкой мозговой оболочкой.
Гидроанэнцефалия - полное или почти полное отсутствие больших полу- шарий при сохранности костей свода черепа и его покровных тканей. Голова при этом нормальных размеров или несколько увеличена. Полость черепа заполнена главным образом ЦСЖ. Продолговатый мозг и мозжечок достаточно развиты. Средний мозг и другие отделы головного мозга могут отсутствовать или представлены рудиментарно. Впервые эта форма порока была описана Ж. Крювелье в 1835 г. под названием «гидроцефалическая анэнцефалия».
Порэнцефалия истинная - наличие в ткани конечного мозга полостей разных размеров, выстланных эпендимой и сообщающихся с желудочковой системой и субарахноидальным пространством.
Порэнцефалия ложная - замкнутые полости в большом мозге, не имеющие эпендимной выстилки и представляющие собой кисты после энцефаломаляции разного происхождения.
Кистозная дисплазия головного мозга, или полипорэнцефалия, - врожденная дисплазия больших полушарий головного мозга, характеризующаяся образованием в нем множественных полостей, обычно сообщающихся с желудочковой системой мозга.
Прозэнцефалия - порок развития, при котором большие полушария моз- га отделяет друг от друга лишь мелкая продольная борозда, поэтому граница между правой и левой половинками конечного мозга нечеткая (встречается с частотой 1:16 000).
Голопрозэнцефалия - порок развития мозга, при котором его большие по- лушария не разделены и имеют вид единой полусферы, а боковые желудочки представлены единой полостью. Часто сочетается с другими врожденными по-
роками. Обычно смерть наступает вскоре после рождения. Может быть проявлением трисомии хромосом 13-15. Пороки конечного мозга сопровождаются различными, порой грубыми, нарушениями строения лица и его костей, в частности цебоцефалией, этмоцефалией и циклопией. Дети с циклопией обычно рождаются мертвыми.
Агирия (лиссэнцефалия) - недоразвитие извилин больших полушарий, при этом поверхность их сглажена (гладкий мозг). При микроскопии выявляются грубое изменение архитектоники коры больших полушарий, отсутствие в ней обычных клеточных слоев. Проявляется выраженным нарушением психомоторного развития, полиморфными судорогами, парезами или параличами. Дети обычно умирают в течение первого года жизни.
Микро- и полигирия - порок, при котором на поверхности больших по- лушарий имеется множество беспорядочно расположенных мелких извилин. Обычно микрогирия проявляется симметрично и сопровождается нарушением послойного строения коры, имеющей не более 4 слоев.
Пахигирия (макрогирия) - укрупнение основных извилин, тогда как вторичные и третичные извилины отсутствуют, борозды при этом выпрямлены, они короткие и неглубокие. Цитоархитектоника коры в таких случаях нарушена. В белом веществе мозга встречаются гетеротопии нервных клеток.
Гипоплазия, или аплазия (агенезия), мозолистого тела - частичное или пол- ное отсутствие мозолистого тела. В случае его аплазии III желудочек мозга остается открытым. Если отсутствует лишь задняя спайка, а само мозолистое тело только укорочено, то это называется гипоплазией.
Синдром Айкарди - гипоплазия мозолистого тела в сочетании с другими пороками, в частности с хориоретинальными аномалиями, при этом характерны спазмы сгибательной мускулатуры или миоклонические приступы, множественные лакунарные очаги в сосудистой и сетчатой оболочках глаз, выявляемые при офтальмоскопии в перипапиллярной зоне. Размеры атрофических хориоретинальных очагов варьируют от небольших, меньше поперечника диска зри- тельного нерва, до диаметра в несколько его поперечников. Часто имеются дизрафические изменения позвоночника. Возможны умственная отсталость, маятникообразный нистагм, аномалии развития глаз (микрофтальм, колобомы зрительного нерва и хориоидальной оболочки, эктазия склеры и др.). Описан синдром только у девочек, это позволяет полагать, что болезнь может быть следствием мутации в Х-хромосоме, которая является летальной при развитии мужского организма. Описал в 1956 г. французский педиатр J. Aicardi.
Микроцефалия (синдром Джакомини) - недоразвитие головного мозга, проявляющееся при рождении уменьшением его массы и размеров (рис. 24.7). Микроцефалия обычно сочетается с уменьшенной окружностью головы (не менее чем на 5 см от средних показателей) и дальнейшим отставанием роста мозгового черепа (микрокрания), при этом швы его могут длительно оставаться открытыми. Кости черепа часто утолщены, в них рано формируются диплоидные каналы, внутричерепное давление не повышено. При микрокрании обычно отмечается соответствующее уменьшение размеров и массы головного мозга - микроцефалия. Морфологическим ее признаком является недоразвитие и неправильное строение больших полушарий при сравнительно нормальной архитектонике мозжечка и ствола мозга. Ребенок с микроцефалией обычно отстает в умственном, а зачастую и в физическом развитии.
Микроцефалия может быть первичной (истинной, генетически обусловленной) и вторичной. Первичная микроцефалия - следствие генетического
Рис. 24.7. Микроцефалия у ребенка 3 лет.
дефекта, наследуемого по аутосомно- рецессивному типу или возникающего в связи с хромосомными аномалиями. Вторичная микроцефалия может быть обусловлена перенесенной внутриутробно инфекцией (краснуха, цитомегаловирусный энцефалит, токсоплазмоз), интоксикаций или асфиксией, травмой мозга. При вторичной микроцефалии в мозге возможны кистозные полости, очаги кровоизлияния и обызвествления. Внешний вид детей с микроцефалией своеобразен и характеризуется диспропорцией между размерами мозгового черепа и лица. Частота микроцефалии среди новорожденных 1:5000. Среди всех случаев олигофрении 11% отмечается у больных с микроцефалией.
Макроцефалия - увеличение массы и объема головного мозга, а вместе с этим и мозгового черепа при рождении, встречается значительно реже микроцефалии. В большинстве случаев сопровождается нарушением расположения мозговых извилин, изменениями цитоархитектоники коры, очагами гетеротопии в белом веществе, при этом обычно отмечаются проявления олигофрении, возможен судорожный синдром. Причиной макроцефалии может быть поражение паренхимы мозга (липоидозы). На краниограммах костные швы не расширены, желудочки мозга нормального или почти нормального размера. Макроцефалию следует дифференцировать от гидроцефалии.
Возможна частичная макроцефалия (увеличение одного из больших полушарий), которая обычно сочетается с асимметрией мозгового черепа. Гемигипертрофия черепа за счет выбухания с одной стороны чешуи височной кости и прилежащих отделов лобной и теменной костей может быть сопряжена с выявляемыми при краниографии углублением и расширением на этой же стороне средней черепной ямки, порозностью крыльев основной кости. В таких случаях гемигипертрофия черепа указывает на вероятность наличия в средней черепной ямке неопухолевого объемного процесса (гематома, гигрома, ксантома, кистозный арахноидит и т.п.) и известна как синдром Дайка.
24.16. ПОРОКИ РАЗВИТИЯ ЖЕЛУДОЧКОВ МОЗГА
Пороки развития вентрикулярной системы обычно проявляются в области ее анатомических сужений. Возможны сужения (стеноз и атрезия) межжелудочковых отверстий, водопровода мозга (сильвиева водопровода), срединной и латеральных апертур IV желудочка мозга. В таких случаях характерно развитие внутренней гидроцефалии, при этом в случае атрезии межжелудочкового
отверстия с одной стороны возникает асимметричная гидроцефалия. Стеноз или атрезия водопровода мозга, а также его расщепление могут наследоваться, передаваясь по аутосомно-рецессивному типу или быть сцеплены с Х-хромосомой. Неполное раскрытие апертур IV желудочка мозга часто сочетается с проявлениями синдрома Денди-Уокера (см. 24.18).
Недостаточность оттока ЦСЖ из желудочковой системы при нарушении проходимости (стенозе) водопровода мозга и апертур IV желудочка мозга проявляется, как правило, развитием внутренней равномерной гидроцефалии, сопровождающейся растяжением, истончением и атрофией ткани мозга. Развитию гидроцефалии нередко сопутствуют и некоторые аномалии основания черепа и верхнего шейного отдела позвоночника: платибазия, симптом Клиппеля- Фейля и др. Возможен также гиперсекреторный или арезорбтивный характер гидроцефалии, обусловленной обычно перенесенным воспалением мозговых оболочек. Частота врожденной гидроцефалии 0,5 на 1000 новорожденных. Подробнее о гидроцефалии см. в главе 20.
24.17. ФАКОМАТОЗЫ
Факоматозы (от греч. phakos - пятно, oma - суффикс, означающий «новообразование», «опухоль», osis - суффикс, означающий «процесс», «болезнь») - группа наследственных заболеваний, при которых наблюдается сочетание поражений нервной системы, кожи и внутренних органов. Характерными проявлениями факоматоза являются участки нарушенной пигментации покровных тканей (гиперпигментированные или депигментированные пятна), шагреневые бляшки, фибромы, папилломы, ангиомы, сочетающиеся с разнообразными неврологическими, психическими, эндокринными и соматическими нарушениями. Для большинства форм факоматозов характерны задержки развития различных функций, прежде всего движений и интеллекта, а также снижение адаптации к экзогенным и эндогенным факторам, факторам социальной среды. В тяжелых случаях наблюдаются олигофрения, атаксия, эпилептические припадки. Описания отдельных вариантов факоматоза появились в конце XIX в.
Морфологической основой факоматозов являются (Архипов Б.А., Карпухина Л.О., 1996) гамартромы, детерминированные нарушениями роста и дифференцировки клеток одного или нескольких зародышевых листков в ранних стадиях эмбриогенеза. Из клеток, которые как бы задержались в своей дифференцировке и находятся в состоянии «перманентной эмбрионизации», об- разуются гамартромы, имеющие склонность к пролиферации и неопластической трансформации. В связи с этим гамартрому расценивают как опухолевидный врожденный порок развития или эмбриональную опухоль с бластоматозными тенденциями (Kousseff B.G. et al., 1990). Гамартромы чаще имеют эктодермаль- ное происхождение и состоят из элементов нервной ткани и кожи. Отсюда и другое название факоматозов - «нейроэктодермальные дисплазии». Они могут сочетаться с мезодермальными и энтодермальными дисплазиями.
Наиболее часто встречаются такие признаки нейроэктодермальной дисплазии, как гипер- и гипопигментированные пятна, пятна цвета «кофе с молоком», фибромы, папилломы, невусы, нейрофибромы, кортикальные и субэпендимальные узелки в ЦНС, факомы, поражения типа «тутовой ягоды» на глазном дне. Среди мезодермальных дисплазий часто встречаются ангиомы, ангиолипомы, аневризмы, эктазии и стенозы сосудов, рабдо- и лейомиомы, дис-
плазии костной ткани и др. Примером энтодермальной дисплазии может быть полипоз различных отделов пищеварительного тракта.
В каталоге наследственных заболеваний V. McKusik (1967) зарегистрированы 54 формы факоматоза. Большинство из них наследуется по аутосомнодоминантному типу.
Нейрофиброматоз, или болезнь Реклингхаузена, встречается чаще других факоматозов (1:4000). В детском возрасте (после 3 лет) появляются множест- венные бледные, желто-коричневые (кофейного цвета) пятна, диаметром от просяного зерна до 15 см и больше, преимущественно на туловище и прок- симальных отделах конечностей; нередко наблюдается генерализованная точечная пигментация или веснушчатость в подмышечных областях. Несколько позже появляются признаки нейрофиброматоза: множественные плотные опухоли разной величины (чаще диаметром 1-2 см), расположенные по ходу нервных стволов (невриномы, нейрофибромы), не сращенные с другими тканями.
Опухоли могут возникать и по ходу черепных нервов (невриномы слухового, тройничного, языкоглоточного нервов). Нередко опухоли растут из ткани спинномозговых корешков и располагаются в позвоночном канале, вызывая сдавление спинного мозга. Опухоли могут локализоваться также в глазнич- ной области, в загрудинном, ретроперитонеальном пространствах, во внутренних органах, вызывая соответствующую разнообразную симптоматику. Часто развивается сколиоз, возможна гипертрофия участков кожи, гипертрофия внутренних органов. В основе заболевания лежат аномалии развития экто- и мезодермы. Возможна астроцитарная гамартрома. Наследуется по аутосомно-доминантному типу. Выделяют 2 формы нейрофиброматоза: классическую, периферическую форму (нейрофиброматоз-1), при которой патологический ген локализован в хромосоме 17, и центральную форму (нейрофиброматоз-2), пато- логический ген расположен в хромосоме 22. Описал болезнь в 1882 г. немецкий патолог F.D. Recklinghausen (1833-1910).
По материалам Института нейрохирургии им. Н.Н. Бурденко РАМН при нейрофиброматозе-1, наряду с периферическими невриномами и нейрофибромами, возможны микроцефалия, пигментированные гамартромы радужки (узелки Лиша), глиомы зрительных нервов (встречаются у 5-10% больных), костные аномалии, в частности дисплазия крыльев основной кости, приводящая к дефекту крыши орбиты и к пульсирующему экзофтальму, односторонние невриномы слухового (вестибулокохлеарного) нерва, внутричерепные опухоли - менингиомы, астроцитомы внутрипозвоночные нейрофибромы, менингиомы, злокачественные опухоли - ганглиобластома, саркома, лейкемия, клинические проявления сирингомиелии.
В случаях нейрофиброматоза-2 часто развивается невринома вестибулокохлеарного черепного нерва, которая при этом заболевании часто бывает двусторонней, возможны менингиома, глиальные опухоли, спинальные невриномы. Возможны также помутнение хрусталика, субкапсулярная лентикулярная катаракта
(Козлов А.В., 2004).
Туберозный склероз (болезнь Бурневилля-Прингла, синдром Бурневилля- Брессау) - глиоз белого вещества мозга, проявляющийся в раннем детстве эпилептическими припадками (в 85%), олигофренией в сочетании с нараста- ющей пирамидной и экстрапирамидной симптоматикой, кожной патологией. В возрасте 4-6 лет на лице в форме бабочки в области носа обычно появляются множественные желто-розовые или коричнево-красные узелки диаметром чуть больше 1 мм - аденомы Прингла, которые обычно признаются аденомами
сальных желез, однако есть мнение и о том, что они представляют собой происходящую из нервных элементов кожи гамартрому.
На носу при этом возможны изменения по типу телеангиэктазий. Часто встречаются участки так называемой шагреневой кожи, пятна кофейного цвета, зоны депигментации, полипы, участки фиброзной гиперплазии, возможны гамартромы языка, фиброзные бляшки на коже лба, волосистой части головы и округлые фибромы (опухоли Коэна) на пальцах ног, реже - рук. Нередко отмечаются диспластические черты, врожденные пороки развития, опухоли сетчатки и внутренних органов (в сердце, почках, в щитовидной и вилочковой железах и пр.).
На глазном дне возможны студенистые образования грязно-желтоватого цвета, напоминающие по форме тутовую ягоду, - глионевромы типа астроцитарной гамартромы, ретинальный факоматоз. Иногда выявляются признаки застоя или атрофии дисков зрительных нервов.
На поверхности мозга наблюдаются единичные или множественные глиоматозные узлы, по цвету несколько светлее окружающего мозга и плотнее его на ощупь, возможна их кальцификация. Узлы могут быть и в белом веществе, подкорковых ганглиях, а также в стволе мозга и в мозжечке.
Встречаются и аномалии развития извилин мозга в виде микро- и пахигирии. Заболевание чаще носит спорадический характер. Бляшки достигают диаметра 5-20 мм. В коре больших полушарий и мозжечка иногда могут быть обнаружены пластинчатые тельца, напоминающие амилоид. Происходит дегенерация клеток коры. При КТ-исследовании головы нередко можно выявить кальцификаты и глиальные узелки в паравентрикулярной области, субэпендимарно вдоль наружных стенок боковых желудочков, в зоне межжелудочкового отверстия Монро, реже - в мозговой паренхиме. На МРТ головного мозга в 60% выявляются гипотеденсивные очаги в одной или обеих затылочных долях, которые расцениваются как участки неправильной миелинизации (Козлов А.В., 2002).
Признается, что заболевание наследуется по аутосомно-доминантному типу с неполной пенетрантностью мутантного гена. Описали его в 1862 г. французский врач D.M. Bourneville (1840-1909) и в 1880 г. английский врач J.J. Pringle
(1855-1922).
Энцефалотригеминальный ангиоматоз Стерджа-Вебера (кожно-мозговой ангиоматоз; синдром Штурге (Стерджа)-Вебера; синдром Вебера-Краббе-Осле-
ра - врожденная мальформация мезодермальных (ангиомы) и эктодермальных элементов, возникшая в процессе эмбриогенеза под воздействием экзогенных и генетически обусловленных причин. Характерна триада: «пламенный» невус, эпилепсия, глаукома. Врожденное крупное сосудистое пятно (невус) обычно ло- кализуется на одной стороне лица по ходу ветвей тройничного нерва. Крупные плоские ангиомы красного или вишневого цвета на лице, бледнеющие при надавли- вании, могут распространяться на кожу волосистой части головы и шею, обычно сопровождаются ангиоматозом мозговых оболочек, чаще в конвекситальной зоне теменно-затылочной области, атрофией мозга и очагами обызвествления в коре больших полушарий. Возможны олигофрения, гемипарез, отставание роста паретичных конечностей, гемианопсия, гидрофтальм. На краниограммах и компьютерных томограммах отмечаются очаги обызвествления, атрофия мозга, расширение субарахноидальных пространств.
Заболевание чаще носит спорадический характер. Возможны случаи наследования как по доминантному, так и по аутосомно-рецессивному типу. На КТ и МРТ обычно наблюдаются проявления атрофии вещества мозга, расшире-
ние желудочков мозга и подоболочечных пространств. Описали заболевание в 1879 г. английские врачи W.H. Sturge (1850-1919) и H.D. Weber (1823-1918).
Атаксия-телеангиэктазия (болезнь Луи-Бар) характеризуется симметричными телеангиэктазиями, появляющимися в возрасте 3-6 лет, особенно на конъюнктиве, коже лица и шеи, обычно распространяющимися на мозговые оболочки, вещество мозга. Кроме того, отмечается повышенная склонность к хроническим воспалительным заболеваниям (синусит, пневмония, бронхоэктазии и пр.) в связи с генетически обусловленным нарушением клеточного и гуморального иммунитета. При первых попытках ребенка к самостоятельной ходьбе выявляются признаки мозжечковой атаксии, которая в дальнейшем имеет нарастающий характер, позднее появляются гиперкинезы по типу миоклонии или атетоза, сухожильная гипорефлексия, дизартрия. Возможно поражение черепных нервов, затруднение произвольных движений глаз (окуломоторная апраксия) . К 12-15 годам возникают нарушения глубокой и вибрационной чувствительности, нарастание атаксии. В поздних стадиях болезни в связи с поражением клеток передних рогов спинного мозга возникают слабость и атрофия мышц, фасцикулярные подергивания. На коже появляются пигментные пятна кофейного цвета, участки гипопигментации, себорейный дерматит. Постепенно раз- вивается атрофия кожи, появление седых волос отмечается уже в школьном возрасте. Характерны задержка психического и физического развития, обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммаглобулинемия, поражение ретикулоэндотелиальной системы (ретикулезы, лимфосаркома и пр.). Прогноз плохой. Причиной смерти чаще являются хронические заболевания бронхов и легких, лимфомы, карциномы.
Наследуется по аутосомно-рецессивному типу с высокой пенетрантностью мутантного гена. Описала болезнь в 1941 г. французская врач D. Louis-Bar.
Цереброретиновисцеральный ангиоматоз (гемангиобластоматоз, болезнь Гиппеля-Линдау) - наследственно-семейный ангиоматоз центральной нервной системы и сетчатки глаз. Характеризуется врожденным недоразвитием капилляров, компенсаторным расширением более крупных сосудов и образованием сосудистых клубочков, ангиом, ангиоглиом. Неврологическая симптоматика может быть разнообразной в связи с возможным поражением больших полушарий, ствола мозга, мозжечка, реже - спинного мозга.
Характерна триада: ангиома сетчатки, ангиомы головного мозга, поликистоз внутренних органов или ангиоретикулема почек. На глазном дне отмечаются резкое расширение и извитость сосудов, желтоватые сосудистые клубочки в сетчатке, позже - экссудат и кровоизлияния в сетчатке, ее отслойка. Часто наблюдаются помутнение стекловидного тела, глаукома, иридоциклит. В результате со временем наступает слепота. Болезнь Гиппеля-Линдау обычно проявляется у больных 18-50 лет.
Первыми симптомами являются признаки ангиоретикулемы мозжечка или сетчатки глаз. При преобладании клинических проявлений ангиоматоза мозжечка болезнь известна как «опухоль Линдау». Ангиоматоз сетчатки обычно рассматривается как «опухоль Гиппеля». Возможны поражения внутренних органов, которые характеризуются аномалиями развития и образованием опухолей: поликистоза почек, феохромоцитомы, гипернефромы, кистозных опухолей поджелудочной железы, печени. Наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Описали болезнь в 1904 г. немецкий офтальмолог Е. Hippel, а в 1925 г. шведский патолог А. Lindau (род. в 1898 г.).
24.18. АНОМАЛИИ И ДЕСТРУКЦИИ НА КРАНИОВЕРТЕБРАЛЬНОМ УРОВНЕ
В зоне перехода черепа в позвоночник часто встречаются краниовертебральные аномалии. Они могут обусловить нарушение кровообращения в позвоночных артериях, расстройство ликворообращения. В результате проявления разнообразных неврологических расстройств, в том числе вестибулярные, мозжечковые симптомы, признаки внутричерепной гипертензии, элементы бульбарного синдрома, в частности нарушения функций черепных нервов бульбарной группы, корешковые симптомы на верхнешейном уровне, признаки пирамидной недостаточности, нарушения чувствительности по проводниковому типу, а также корешковые симптомы на верхнешейном уровне. Могут выявляться многообразные костные аномалии, проявления дизрафического статуса: базилярное вдавление, выстояние верхушки зубовидного отростка выше линий Чемберлена и де ля Пети, ассимиляция атланта (синдром Ольенека), феномен проатланта и др. Для краниовертебральных аномалий характерны короткая шея, низкая граница роста волос на шее, шейный гиперлордоз; возможны асимметрия лица, гипоплазия нижней челюсти, готическое нёбо, расширение позвоночного канала на уровне верхних шейных позвонков, кифосколиоз позвоночника, расщепление дужек позвонков, деформация стоп по типу «стопы Фридрейха».
Врожденные аномалии развития на краниовертебральном уровне характеризуются дефектами развития затылочной кости и расположенных в задней черепной ямке структур и верхнего отдела позвоночника и спинного мозга. К ним относятся синдромы Денди-Уокера и синдром Киари.
Синдром Денди-Уокера представляет собой врожденный порок развития ка- удального отдела ствола и червя мозжечка, ведущий к неполному раскрытию срединной (Мажанди) и латеральных (Лушки) апертур IV желудочка мозга. Проявляется признаками гидроцефалии, а нередко и гидромиелии. Последнее обстоятельство в соответствии с гидродинамической теорией Гарднера может обусловить развитие сирингомиелии, сирингобульбии. Синдром Денди-Уокера характеризуется проявлениями функциональной недостаточности продолговатого мозга и мозжечка, симптомами гидроцефалии, внутричерепной гипертензии. Уточ- няется диагноз с помощью визуализирующих мозговую ткань методов - КТ- и МРТ-исследований. Выявляются признаки гидроцефалии, в частности выраженное расширение IV желудочка мозга, при МРТ-исследовании можно выявить деформацию указанных структур мозга. Описали синдром в 1921 г. американские нейрохирурги W. Dandy (1886-1946) и A. Walker (род. в 1907 г.).
Синдром Киари (синдром Арнольда-Киари-Соловцева, или синдром церебелломедулярного уродства) - порок развития субтенториальных структур ромбовидного мозга, проявляющийся опущением ствола мозга и миндалин мозжечка в большое затылочное отверстие. Нередко сочетается с аномалиями костей основания черепа и верхних шейных позвонков (платибазией, базилярной импрессией, ассимиляцией атланта, синдромом Клиппеля-Фейля), с проявлениями дизрафического статуса, в частности с сирингомиелией, сирингобульбией. При синдроме Киари могут возникать ущемление продолговатого мозга, структур мозжечка, верхних шейных сегментов спинного мозга, окклюзия ликворных путей, что ведет к бульбарным, мозжечковым и проводниковым симптомам, к окклюзионной гидроцефалии. Описали синдром в
1894 г. немецкий патолог J. Arnold (1835-1915) и в 1895 г. австрийский патолог H. Chiari (1851-1916).
В настоящее время, основываясь на результатах МРТ-сканирования, некоторые авторы выделяют два варианта синдрома Киари.
Мальформация I типа (Киари I) характеризуется смещением миндалин мозжечка до уровня большого затылочного отверстия. Возможно опущение продолговатого мозга, его удлинение и передняя компрессия продолговатого мозга зубовидным отростком, сужение IV желудочка мозга и большой затылочной цистерны, ликвородинамические расстройства, признаки недоразвития и атипичного строения артерий вертебрально-базилярного бассейна. В неврологическом статусе возможны глазодвигательные, кохлеарные и вестибуломозжечковые, бульбарные, а также проводниковые двигательные и сегментарные двигательные и чувствительные нарушения. Отсутствие неврологических симптомов, однако они могут проявиться позже (иногда на 3-4 десятилетии жизни, что свидетельствует о переходе процесса в мальформацию II типа.
При мальформации II типа (Киари II) отмечается протрузия в большое затылочное отверстие миндалин и червя мозжечка, структур продолговатого мозга, который принимает S-образную форму. Характерны спастический тетрапарез, боли в затылочной области и шее, мозжечковая атаксия, вертикальный «бьющий» вниз нистагм, элементы бульбарного синдрома, признаки сирингомиелии, проявления гидроцефалии, проводниковые нарушения.
Неврологическая симптоматика при синдроме Арнольда-Киари может проявляться с 5-7 лет, иногда позже, возможно в 30-40-летнем возрасте, и имеет прогредиентное течение. Проявления аномалии Арнольда-Киари нередко сочетаются с краниовертебральной костной аномалией (базилярная импрессия, ассимиляция атланта, краниостеноз по типу скафокрании и др.). В диагностике синдрома Киари и определении его типа особенно ценной обычно является информация, полученная при МРТ головного мозга и краниовертебральной области, а также при транскраниальной допплерографии (Крупина Н.Е., 2003).
Симптом Бабчина - атрофия заднего полукольца большого затылочного отверстия и внутреннего гребешка затылочной кости. Выявляется при кранио- графии, выполненной в задней полуаксиальной проекции. Описан симптом отечественным нейрохирургом И.С. Бабчиным при опухолях краниовертебральной локализации.
24.19. НЕКОТОРЫЕ ВРОЖДЕННЫЕ ИЛИ РАНО ПРОЯВЛЯЮЩИЕСЯ ФОРМЫ ПОРАЖЕНИЯ ДВИГАТЕЛЬНОЙ СФЕРЫ
24.19.1. Детские церебральные параличи
Детские церебральные параличи (ДЦП) - гетерогенная группа синдромов, которые являются следствием повреждений мозга, возникших во внутриутробном, интранатальном (во время родов) и раннем постнатальном периодах. Характерная особенность ДЦП - нарушение моторного развития ребенка, обусловленное прежде всего аномальным распределением мышечного тонуса и нарушением координации движений (парезы, параличи, атаксия, гиперкинезы). Отмеченные
двигательные расстройства могут сочетаться с приступами эпилепсии, задержкой развития речи, эмоционального и интеллектуального развития. Иногда расстройства движений сопровождаются и изменением чувствительности.
Важной особенностью ДЦП является отсутствие прогрессирования и возможная, хотя и слабо выраженная, тенденция к восстановлению имеющихся признаков патологии нервной системы.
Частота ДЦП, по разным данным, составляет 2,5-5,9 на 1000 новорожденных. По данным Московской детской консультативной неврологической поликлиники, в 1977-1978 гг. она составила 3,3 на 1000 детского населения. Частота ДЦП в группе детей, родившихся с массой тела менее 1500 г, составляет 5-15% (Aziz K. et al., 1994). Согласно данным К.А. Семеновой (1994), ДЦП является причиной 24% случаев детской неврологической инвалидности.
Этиология . Этиологические факторы разнообразны: заболевания (краснуха, цитомегалия, грипп, токсоплазмоз и др.) и токсикозы у матери во время беременности, аномалии родовой деятельности, акушерские операции и трав- матические поражения, кровоизлияния в мозг, асфиксии во время родового акта, несовместимость крови матери и плода, травмы и болезни (менингиты, энцефалиты) у ребенка в раннем послеродовом периоде. Возможно сочетание нескольких вредных факторов.
Причинами врожденного ДЦП могут быть генетические детерминированные аномалии формирования мозга (дисгенезии мозга), возникающие на разных этапах его развития. Они являются причиной 10-11% всех случаев спастических форм ДЦП. Кроме того, причиной ДЦП могут быть сосудисто-мозговые нарушения у плода или новорожденного ребенка, в частности гипоксически-ишемическая энцефалопатия, ишемические и геморрагические инсульты, внутричерепные гематомы.
Патогенез. Патогенные факторы, действующие во время эмбригенеза, вызывают аномалии развития мозга. На более поздних этапах внутриутробного развития возможно замедление процессов миелинизации нервной системы, нарушение дифференциации нервных клеток, патология формирования межнейронных связей и сосудистой системы мозга. При несовместимости крови матери и плода по резус-фактору, системе АВ0 и другим антигенам эритроцитов в организме матери вырабатываются антитела, вызывающие гемолиз эритроцитов плода. Непрямой билирубин, образовавшийся в процессе гемолиза, оказывает токсическое действие на нервную систему, в частности на структуры стриопаллидарной системы.
У плодов, перенесших внутриутробную гипоксию, к моменту рождения защитные и адаптационные механизмы оказываются недостаточно сформированными, существенное значение могут иметь асфиксия и черепно-мозговая травма во время родов. В патогенезе поражений нервной системы, развивающихся во время родов и постнатально, главную роль играют гипоксия плода, ацидоз, гипогликемия и другие метаболические нарушения, ведущие к отеку мозга и вторичным расстройствам мозговой гемодинамики и ликвородинамики. Существенное значение в патогенезе ДЦП придается иммунопатологическим процессам: мозговые антигены, образующиеся при деструкции нервной системы под влиянием инфекций, интоксикаций, механических повреждений мозговой ткани, могут привести к появлению соответствующих антител в крови матери, что негативно сказывается на развитии мозга плода.
Патоморфологическая картина. Патоморфологические изменения нервной системы при ДЦП многообразны. У 30% детей имеются аномалии развития
мозга - микрогирия, пахигирия, гетеротопия, недоразвитие полушарий и др. Возможны дистрофические изменения мозга, глиоматоз, рубцы, порэнцефалия или кистозные полости в мозге, участки демиелинизации проводящих путей или атрофии коры больших полушарий в связи с травматическим поражением, кровоизлиянием в мозг, внутричерепной гематомой, гипоксией, возникшими в процессе родового акта или токсическим, инфекционно-аллергическим, травматическим поражением мозга во внутриутробном или раннем постнатальном периодах.
Классификация. Предлагаются разные клинические классификации ДЦП. Мы приводим одну из классификаций, получивших широкое признание.
Таблица 24.1. Синдромы (формы) ДЦП (Miller G., 1998)
Преобладающими являются спастические формы, остальные встречаются значительно реже.
Клинические проявления. Возникший дефект в мозге не только негативно сказывается на состоянии новорожденного ребенка, но и препятствует его нормальному развитию, прежде всего развитию двигательной системы, речи и когнитивных функций. Клиническая картина в таких случаях может варьировать в больших пределах. Важно помнить, что патологическая постуральная активность, проявления повышения мышечного тонуса нередко становятся отчетливыми только к 3-4 месяцу жизни ребенка, а иногда и позже. Для относительно ранней диагностики ДЦП имеет значение динамическое наблюдение за детьми, особенно с неблагополучным акушерским анамнезом, учет при этом динамики врожденных безусловных рефлексов, последовательности характера изменений мышечного тонуса становления реакций выпрямления и равновесия.
По преобладанию тех или иных неврологических и психических функций Л.О. Бадалян (1984) выделял следующие варианты ДЦП.
1. Спастическая диплегия (синдром Литтля) - наиболее часто встречающаяся форма ДЦП. Характеризуется тетрапарезом с вовлечением в процесс мышц лица, языка, глотки, при этом особенно выражены двигательные расстройства в нижних конечностях (проявления нижнего спастического парапареза с преобладанием напряжения приводящих мышц бедер и мышц-разгибателей голени и сгибателей стоп. Если ребенок лежит, ноги у него вытянуты, при попытке поставить на пол) ноги у него перекрещиваются, он опирается не на всю стопу, а только на переднюю ее часть. Ноги при этом выпрямлены и ротированы внутрь. При попытке ходить с посторонней помощью ребенок совершает танцующие движения, ноги его «перекрещиваются», тело поворачивается в сторону ведущей ноги. Нередко выраженность парезов асимметрична, при этом различие в возможности активных движений особенно отчетливо проявляется в руках.
На фоне диплегии могут быть хореоатетоидные гиперкинезы, в которые вовлекаются прежде всего мимические мышцы и мышцы дистальных отделов рук. Дети тяжело переживают наличие двигательных нарушений, неохотно
вступают в контакт со здоровыми детьми, лучше чувствуют себя в коллективе, состоящем из детей с подобными болезнями.
2. Двойная гемиплегия - двусторонняя гемиплегия или, чаще, гемипарез, при котором руки страдают в большей степени, чем ноги, или они поражены приблизительно в равной мере. Возможна асимметрия выраженности парезов, при этом тонус мышц высокий, имеется сочетание спастики и ригидности обычно с преобладанием последней. Реакции равновесия развиты недостаточно. Почти всегда выражены элементы псевдобульбарного паралича, в связи с чем затруднены жевание и глотание, речь. Нередко отмечаются судорожные пароксизмы, микроцефалия. Эта форма ДЦП обычно сопровождается наиболее значительными проявлениями олигофрении.
3. Спастическая гемиплегия характеризуется соответствующими двигательными нарушениями преимущественно на одной стороне. Нередко двигательные расстройства более выражены в руке, она согнута во всех суставах, кисть у детей раннего возраста сжата в кулак, в более позднем возрасте имеет форму «руки акушера». Нередко возникают фокальные эпилептические припадки по типу Джексона. С помощью визуализирующих методов исследования (КТ, МРТ) в одном из полушарий мозга обычно выявляют кисту, рубцовые процессы или проявления порэнцефалии. Развитие интеллекта может оказаться близким к нормальному.
4. Гиперкинетическая форма характеризуется преимущественным поражением структур стриопаллидарной системы. Мышечный тонус изменчив, часто колеблется между гипотонией и нормотонией. На этом фоне возникают перемежающиеся мышечные спазмы, приступы повышения мышечного тонуса по пластическому типу. Активные движения в таких случаях неловкие, сопровож- даются излишними двигательными реакциями преимущественно атетоидного характера, при этом гиперкинезы могут быть преимущественно в дистальных или проксимальных отделах конечностей, мышцах шеи, в мимической мускулатуре. Гиперкинезы возможны по типу атетоза, хореоатетоза, хореи, торсионной дистонии. Часто наблюдаются расстройства речи (подкорковая дизартрия). Психическое развитие страдает меньше, чем при других формах ДЦП. Эта форма ДЦП обычно обусловлена иммунной несовместимостью крови плода и матери.
5. Мозжечковая форма характеризуется атаксией, обусловленной главным образом поражением мозжечка и его связей. Может сочетаться с нистагмом, атонически-астатическим синдромом, признаками умеренного спастического пареза в связи с вовлечением в процесс корково-подкорковых структур мозга.
Лечение . Лечение, точнее, абилитацию 1 больного с ДЦП следует начинать как можно раньше, при этом оно должно быть комплексным. В раннем воз- расте мозг ребенка пластичен и обладает значительными компенсаторными возможностями. Абилитация, начатая в период формирования статических и локомоторных функций, дает наиболее существенные результаты. Раннее обучение сенсомоторным навыкам с условнорефлекторным их закреплением способствует своевременному развитию моторики. Кроме того, в раннем возрасте спастические явления выражены еще нерезко, отсутствуют стереотипные патологические позы, деформации, контрактуры, вследствие чего легче вырабатываются двигательные навыки.
1 Абилитация - создание возможностей для развития отсутствовавших ранее видов деятельности.
Важной частью комплексного лечения ДЦП являются ортопедические мероприятия, профилактика контрактур. Для придания физиологического положения отдельным частям тела широко используются лонгеты, туторы, шины, валики, воротники и пр. Ортопедические укладки чередуются с лечебной гимнастикой, массажем, физиотерапией, при этом лечебные мероприятия должны способствовать торможению патологической тонической рефлекторной актив- ности, нормализации на этой основе мышечного тонуса, облегчению произвольных движений, развитию последовательных возрастных двигательных навыков ребенка.
Из лекарственных средств в процессе лечения ДЦП применяются фармакопрепараты, улучшающие метаболические процессы в мозге, - глутаминовая кислота, липоцеребрин, церебролизин, препараты из группы ноотропов, витамины группы В, ацефен и др. По показаниям применяют миорелаксанты, при этом средством выбора может быть ботокс (ботулинический токсин). Есть положительный опыт (Белоусова Е.Д., Темин П.А. и др., 1999) его введения в двуглавую мышцу плеча, а также в сгибатели и разгибатели кисти для уменьшения мышечного тонуса и пронаторной установки предплечья; положительный эффект дало применение ботокса теми же авторами для ликвидации динамической контрактуры в голеностопном суставе. Применяются также препараты, действие которых направлено на подавление гиперкинезов, противосудорожные средства, ангиопротекторы, антиагреганты и седативные средства.
В последние годы разрабатываются методы соматосенсорной стимуляции. Для этого предлагается, в частности, ношение космического нагрузочного костюма «Пингвин» или его модификации «Адели». Применение нагрузочного костюма способствует исправлению положения центра тяжести тела больного и нормализации позы стояния. Предполагается (Яворский А.Б. и др., 1998), что при таком лечении может происходить перестройка нервных связей в полушариях мозга и изменение при этом межполушарных взаимоотношений.
24.19.2. Спастическая семейная параплегия Штрюмпелля
Хроническая прогрессирующая семейная болезнь подробно описана в 1886 г. немецким врачом А. Штрюмпеллем (Stumpell A., 1853-1925). В настоящее время она расценивается как группа заболеваний, характеризующаяся генетической гетерогенностью и клиническим полиморфизмом. Болезнь наследуется как по аутосомно-рецессивному, так и по доминантному типу.
Патогенез не изучен.
Патоморфологическая картина. Отмечается симметричная дегенерация в церебро-спинальных проводящих путях, постепенно прогрессирующая и рас- пространяющаяся снизу вверх. Иногда она сопровождается дегенеративными изменениями в нежном пучке Голля и спинномозговых трактах. Возможны демиелинизация нервных волокон в ножках мозга, глиоз и уменьшение количества клеток в экстрапирамидных структурах ствола.
Клинические проявления . Обычно на втором десятилетии жизни появляются утомляемость ног, повышение в них мышечного тонуса и сухожильных рефлексов. Позже возникают клонусы стоп, стопные патологические знаки. Со временем признаки нижнего спастического парапареза нарастают, при этом спастическое состояние мышц преобладает над выраженностью мышечной
слабости. В течение многих лет больные сохраняют способность к самостоятельному передвижению. Походка у них спастическая парапаретическая. Изза выраженности напряжения приводящих мышц бедер больные иногда при ходьбе перекрещивают ноги. В далеко зашедшей стадии заболевания возможны защитные рефлексы, признаки спинномозгового автоматизма, контрактуры голеностопных суставов. Элементы спастичности могут по мере развития заболевания проявиться в руках, в мышцах плечевого пояса. Возможно снижение вибрационной чувствительности на ногах. Другие виды чувствительности, трофика тканей и функции тазовых органов обычно не страдают. Возможны деформация стоп (стопа Фридрейха), легкая мозжечковая недостаточность, миокардиопатия, снижение когнитивных функций.
Лечение . Патогенетическая терапия не разработана. В качестве симптома- тических средств широко применяются миорелаксанты (мидокалм, скутамил, баклофен и др.).
24.20. АНОМАЛИИ И ВТОРИЧНЫЕ ДЕФОРМАЦИИ ПОЗВОНОЧНИКА
К краниовертебральным костным аномалиям относится симптом Ольеника - окципитализация I шейного позвонка (атланта) - сращение его (ассимиляция, конкресценция) с затылочной костью. Этот симптом может сопровождаться признаками краниовертебральной патологии, вертебрально-базилярной сосудистой недостаточностью, нарушениями ликвородинамики. На спондилограммах иногда выявляется феномен проатланта - наличие элементов дополнительного («затылочного») позвонка в виде рудиментов передней дуги, тела, бокового отдела или задней дуги. Чаще они находятся в состоянии слияния с затылочной костью, атлантом, верхушкой зубовидного отростка II шейного (осевого) позвонка, однако могут сохраняться и в виде свободных косточек, расположенных в связочном аппарате между затылочной костью и атлантом.
Проявлением врожденного костного дефекта является аномалия Кимерли. Борозда позвоночной артерии на тыльной стороне боковой массы атланта оказывается преобразованной в частично или полностью замкнутый канал в связи с образованием над ней костного мостика. Это может обусловить сдавливание проходящей через этот канал позвоночной артерии и развитие вертебральнобазилярной сосудистой недостаточности, которая иногда проявляется с юных лет. Описал патологию в 1930 г. M. Kimerly.
Подвывих и расклинивание атлантоосевого сустава, или сустава Крювелье, обусловлены дефектностью его формирования и нередким внедрением в него свободных фрагментов проатланта, что ведет к развитию в этом суставе признаков деформирующего артроза. Возможно проявление болезни Дауна, болезни Моркио, ревматоидного полиартрита, травмы шеи. К развитию подвывиха атлантоосевого сустава предрасполагают слабость связочного аппарата шеи, гипоплазия зубовидного отростка, а также наличие так называемой суставоподобной щели между зубовидным отростком и телом II шейного позвонка. Больные обычно отмечают боли в шее и ограничение подвижности головы, при ее поворотах болезненность и хруст. Неврологические расстройства возникают в результате нестабильности в атлантоосевом суставе и нередко провоцируются легкой травмой шеи, при этом возможно смещение атланта вперед и сдавление верхнешейного отдела спинного мозга.
В случаях поражения двух верхних шейных позвонков при туберкулезной инфекции (болезнь Руста) , сифилисе, ревматизме, метастазах раковой опухоли на спондилограммах отмечаются соответствующие этиологическому фактору изменения в верхних шейных позвонках, иногда и в затылочной кости (см. главу 29).
Болезнь Гризеля (кривошея Гризеля) - спондилоартрит верхнешейного отдела. Возникает чаще у детей на фоне инфекционных заболеваний, иногда является осложнением синусита. Характерно поражение сочленения между атлантом и зубом осевого позвонка. Проявляется резкой болью и болезненностью в верхней шейной области, а также противоболевой контрактурой мышц, прикрепляющихся к атланту. Характерна стойкая спастическая кривошея, при которой голова наклонена в сторону очага поражения и слегка ротирована в противоположном направлении (см. главу 29).
Синдром осевого позвонка является следствием аномалии развития зубовидно- го отростка осевого позвонка, служит основой формирования синдрома зубовид- ного отростка, который оказывается не сращенным с его телом и представлен самостоятельной зубовидной костью (os odontoideum) . Эта кость свободно смещается при наклонах головы, сужая таким образом позвоночный канал, что может обусловить развитие на верхнешейном уровне компрессионной миелопатии; в таком случае возможно возникновение проводниковой симптоматики и дыхательных нарушений, а также появление признаков деформирующего артроза преимущественно в боковых атлантоосевых сочленениях с увеличением их суставных поверхностей за счет костных разрастаний с постепенной миграцией суставов вперед и вниз, т.е. с формированием краниовертебрального спондилолистеза. Могут возникать и проявления сосудистой вертебральнобазилярной недостаточности.
Синдром Клиппеля-Фейля (синдром короткой шеи) представляет собой врожденные аномалии и сращение шейных позвонков, часто сочетается с синдромом Ольеника. Возможна неполная дифференцировка шейных позвонков и уменьшение их числа, иногда количество их не превышает четырех. В клинической картине характерна триада: короткая шея («человек без шеи», «шея лягушки»), низкая граница роста волос на шее, значительное ограничение подвижности головы. В тяжелых случаях подбородок упирается в грудину, мочки ушных раковин касаются надплечий, иногда - кожные складки идут от ушных раковин к плечам. Может сочетаться с гидроцефалией, элементами бульбарного синдрома, вертебрально-базилярной сосудистой недостаточностью, проводниковы- ми нарушениями, высоким стоянием лопаток, проявлениями дизрафического статуса. По данным рентгенологических исследований, выделяют две крайние формы синдрома Клиппеля-Фейля: 1) атлант слит с другими шейными позвонками, общее количество которых в связи с этим уменьшено, обычно их не более 4; 2) признаки синдрома Ольеника и синостоз шейных позвонков, высота их тел снижена. Нередко сочетается с платибазией, возможны другие пороки развития. Описали синдром в 1912 г. французские невропатологи М. Klippеl (1858-1942) и А. Feil (род. в 1884 г.).
Мышечная врожденная кривошея - укорочение грудино-ключично-сосцевидной мышцы вследствие очагового ее фиброза, в результате чего голова наклонена в пораженную сторону. Причина возникающего при этом синдрома замещения участка мышцы соединительной тканью неизвестна.
Конкресценция позвонков - сращение соседних позвонков в связи с аномалией их развития или вследствие туберкулезного спондилита, болезни Бехтерева, посттравматического спондилеза и других патологических процессов.
Платиспондилия - расширение и уменьшение высоты тел позвонков в свя- зи с развитием в них дегенеративных или некротических процессов.
Генерализованная платиспондилия (синдром Дрейфуса) - энхондральный дизостоз, проявляющийся обычно на втором году жизни ребенка (когда он начинает ходить) болью в спине и слабостью фиксирующего позвоночник связочного аппарата с последующим развитием кифоза или кифосколиоза. Характерны короткие шея и туловище при относительно длинных конечностях, гипотрофия и чрезмерная растяжимость мышц, разболтанность суставов. На спондилограмме видна множественная платиспондилия, при этом высота тел позвонков может быть уменьшена в 2-3 раза, расширение пространств между телами позвонков, возможны уменьшенные размеры костей таза и крестца, врожденный вывих бедра или бедер. Описал синдром в 1938 г. французский врач J.R. Dreyfus.
Остеопатия тела позвонка, проявляющаяся обычно у детей в 4-9-летнем возрасте, известная как синдром плоского позвонка (болезнь Кальве). На спон- дилограммах отмечается остеопороз центральной части тела позвонка, уплотнение замыкательных пластинок с последующим прогрессивным его уплощением (платиспондилия) до 25-30% его исходной высоты. Уплощенный позвонок отделен от соседних расширенными межпозвонковыми дисками (см. главу 29).
Патологический лордоз и кифоз позвоночника. Позвоночный столб в норме имеет физиологические изгибы. Изгиб вперед (лордоз) обычно имеет место на шейном и поясничном уровнях, изгиб назад (кифоз) - на грудном. Чрезмерная выраженность лордоза ведет к увеличению нагрузки на задние отделы межпозвонковых дисков, а также на межпозвонковые суставы, в которых в таких случаях могут развиваться дистрофические явления. При цервикалгии или люмбалгии на соответствующем уровне отмечается уплощение лордоза, а иногда и трансформация его в кифоз. При миопатиях обычно усиление выраженности поясничного лордоза.
Патологический кифоз характерен для туберкулезного спондилита, он может возникать при цервикалгии или люмбалгии у больных с остеохондрозом позвоночника, резко выражен при юношеском кифозе, синдроме Линдемана, синдроме Шейермана (см. главу 29).
Если лордоз и кифоз могут быть физиологическими, то сколиоз - стойкий изгиб позвоночника в сторону - всегда признак отклонения от нормы. Выделяются 3 степени сколиоза: I - выявляется только при функциональных пробах, в частности при наклонах туловища в сагиттальной и фронтальной плоскостях; II - определяется при осмотре стоящего больного, исчезает при подтягивании на выпрямленных руках, на параллельных брусьях или на спинках двух стульев, а также в положении лежа на животе; III - стойкий сколиоз, не исчезающий при подтягивании на гимнастической стенке и т.п. и в положении лежа на животе. Рентгенологически обнаруживаемое расширение щелей между телами позвонков на выпуклой стороне искривления позвоночника при сколиозе нередко именуется признаком Кона - по имени отечественного ортопеда И.И. Кона (род. в 1914 г.), описавшего этот признак как проявление прогрессирующего сколиоза. Сочетание кифоза и сколиоза называется кифосколиозом.
Синдром ригидного позвоночника - миопатический синдром, сочетающийся с фиброзом и укорочением аксиальной мускулатуры, особенно разгибателей позвоночника, при этом нарушается сгибание головы и туловища, обычен сколиоз
грудного отдела позвоночника с контрактурами проксимальных суставов конечностей. На ЭМГ отмечаются признаки поражения клеток передних рогов спинного мозга и мышц. Характерны мышечная слабость, миогипотрофии, признаки кардиомиопатии и изменение активности креатинфосфокиназы. Наследуется по аутосомно-рецессивному или по Х-сцепленному рецессивному типу. Описали в 1865 г. английский врач V. Dubowitz, а под названием «врожденный артрогрипоз позвоночника» в 1972 г. - отечественная невропатолог Ф.Е. Горбачева.
Деформации позвоночника могут возникать в процессе инволюции. Такие изменения формы позвоночника наблюдаются, в частности, при синдроме Форестье, проявляющемся у лиц 60-80-летнего возраста, при этом характерна круглая старческая спина.
При чрезмерном поясничном лордозе вследствие давления остистых отростков друг на друга возможна их деформация (синдром Бострупа, «целующийся» остистый отросток). Проявляется болью в поясничной области при разгибании позвоночника. На спондилограммах выявляются ложные суставы между уплощенными остистыми отростками.
Уплощение тела позвонка и заострение при этом его передней части представляют собой одно из проявлений остеохондродистрофии, известное как деформация Моркио-Брейлсфорда.
О последних трех клинических феноменах см. также в главе 29.
24.21. ДИЗРАФИИ ПОЗВОНОЧНИКА И СПИННОГО МОЗГА, СПИННОМОЗГОВЫЕ ГРЫЖИ
Спинальная дизрафия представляет собой порок развития, связанный с не- полным закрытием тканей мезодермального и эктодермального происхождения вдоль срединного шва (от греч. rhaphe - шов) - средней линии позвоночника. Проявлениями спинальной дизрафии являются расщепление дуг позвонков (spina bifida) и сагиттально расположенных мягких тканей, а также возникающие при этом различные варианты спинномозговых грыж, иногда дермоидные кисты, липомы, синдром «жесткой» конечной нити.
Дизрафия позвоночника и спинного мозга в зависимости от степени их недоразвития имеет следующие варианты: 1) spina bifida occulta; 2) spina bifida complicata; 3) spina bifida anterior; 4) спинномозговые грыжи: менингоцеле, ме- нингорадикулоцеле, миеломенингоцеле, миелоцистоцеле; 5) рахисхизис частичный и полный.
Скрытая расщелина позвоночника - spina bifida occulta (от лат. spina - ость, bifidus - надвое разделенный). Наиболее частая форма аномалии позвоночни- ка - расщепление дуг позвонков (spina bifida occulta). Незаращенными могут быть 1-2 позвонка, но иногда и большее их количество. Концы незаращенных дуг нередко вдавливаются в просвет позвоночного канала и вызывают компрессию твердой мозговой оболочки, субдурального пространства и корешков конского хвоста, при этом костный дефект прикрыт неизмененными мягкими тканями. Такая форма аномалии выявляется при спондилографии, чаще на нижнепоясничном - верхнекрестцовом уровнях. В зоне расщепления дуги или нескольких дуг позвонков иногда отмечается втянутость и атрофия кожи или же припухлость тканей, рубцы, пигментация, возможен гипертрихоз -
симптом Фавна. Наличие spina bifida occulta может предрасполагать к развитию болевого синдрома, иногда - синдрома Лермитта, сопровождающегося ощущением по типу прохождения электрического тока вдоль позвоночника при постукивании по остистому отростку аномального или поврежденного позвонка.
Полный рахисхизис - выраженная дизрафия, проявляющаяся расщеплением не только дуг и тел позвонков, но и прилежащих к ним мягких тканей. Через расщелину в мягких тканях сразу же после рождения ребенка может быть виден спинной мозг. Грыжевого выпячивания тканей при этом нет. Тела позвонков в вентральной части расщелины могут срастаться. Возможны пороки развития и других позвонков, ребер. Встречаются парциальные, субтотальные и тотальные формы дизрафии.
Spina bifida anterior - незаращение тел позвонков. Встречается редко и в основном является случайной находкой на спондилограммах, однако может сочетаться с другими дефектами развития.
Spina bifida complicata - незаращение дуг позвонка в сочетании с опухоле- видными разрастаниями, представляющими собой всего лишь жировую или фиброзную ткань, расположенную под кожей и заполняющую костные дефекты дуг позвонков, срастаясь при этом с мозговыми оболочками, корешками и спинным мозгом. Локализуется чаще на пояснично-крестцовом уровне позвоночного столба.
Спинномозговые грыжи, возникающие в связи с незаращением дуг позвонков и расщеплением мягких тканей, представляют собой врожденные грыжевые выпячивания содержимого позвоночного канала (рис. 24.8): менингоцеле - грыжевое выпячивание из мозговых оболочек, заполненное ЦСЖ; менингорадикулоцеле - грыжа, состоящая из мозговых оболочек, спинальных корешков и ЦСЖ; миелорадикуломенингоцеле - грыжа, включающая структуры спинного мозга, спинномозговые корешки, мозговые оболочки и ЦСЖ; миелоцистоцеле - грыжевой мешок, содержащий участок спинного мозга с признаками гидромиелии.
Диагностика . При спинномозговых грыжах диагностика не представляет трудности. О характере содержимого грыжевого мешка можно судить на ос-
Рис. 24.8. Ребенок со спинномозговой грыжей (миеломенингоцеле) и сопутствующей гидроцефалией.
новании изучения неврологического статуса. Уточнение диагноза может быть обеспечено с помощью спондилографии и МРТ-исследования, при этом надо иметь в виду, что окостенение крестца происходит лишь приблизительно к 12 годам жизни.
Лечение спинномозговых грыж. Возможно только хирургическое лечение. При быстром увеличении, истончении и изъязвлении покровных тканей грыжевого выпячивания, угрожающих его разрывом, а также наличием ликворного свища, показана срочная операция. В противном случае возможно развитие менингита, менингомиелита, менингомиелоэнцефалита. Противопоказанием к операции может быть воспаление тканей позвоночного канала, выраженные неврологические расстройства. Вопрос об операции должен решаться совместно педиатром, невропатологом и нейрохирургом.
Грыжевое выпячивание выделяется из мягких тканей, стенка его вскрывается. Если в полость грыжи выпячиваются корешки и ткань самого спинного мозга, то их по возможности с соблюдением максимальной осторожности выделяют из сращений и перемещают в просвет позвоночного канала. После этого грыжевое выпячивание иссекают и последовательно послойно зашивают мягкие ткани. При больших дефектах иногда производят перемещение мышц и апоневроза из прилежащих областей для полноценного закрытия дефекта и предупреждения повторных выпячиваний. Если в грыжевой мешок попадает спинной мозг, как правило, возможна лишь паллиативная операция.
При лечении спинномозговых грыж следует учитывать тот факт, что они нередко сочетаются с гидроцефалией. В этих случаях, помимо удаления грыжевого выпячивания, показана шунтирующая операция - люмбоперитонеостомия.
24.22. АНОМАЛИИ СПИННОГО МОЗГА
Диастематомиелия - разделение спинного мозга по длиннику на две части костной, хрящевой или фиброзной перемычкой. Для такой формы аномалии нет облигатных признаков, так как имеющиеся симптомы возможны и при других пороках развития позвоночника и спинного мозга. Тем не менее диастематомиелия может сопровождаться кожными проявлениями, отклонением от нормы в состоянии опорно-двигательного аппарата и неврологическими расстройствами.
При внешнем осмотре пациента с диастематомиелией вдоль оси позвоночника в зоне расщепления спинного мозга могут быть видны участки гипертрихоза, пигментные пятна кофейного или темно-коричневого цвета, ангиомы, а также втянутые участки рубцово-измененной кожи.
Изменения опорно-двигательного аппарата возможны уже в раннем детстве. Возможны, в частности, деформации стоп. Слабость одной или обеих ног, асимметрия мышц нижних конечностей, гипотрофия отдельных мышц или мышечных групп, слабость мышц ног и тазового пояса. Нередко у детей с раннего возраста выявляются сколиоз и другие формы деформации позво- ночника.
Из неврологических симптомов могут отмечаться асимметрия или отсутствие сухожильных рефлексов, чаще пяточного (ахиллова) или коленного, снижение чувствительности, признаки нарушения вегетативной иннервации.
Иногда значительные по степени выраженности признаки нижнего парапареза сочетаются с расстройством функций тазовых органов, при этом могут быть императивные позывы на мочеиспускание, ночное недержание мочи.
Амиелия - полное отсутствие спинного мозга, при этом сохранены твердая мозговая оболочка и спинномозговые ганглии. На месте спинного мозга возможен тонкий фиброзный тяж.
Дипломиелия - удвоение спинного мозга на уровне шейного или поясничного утолщения, реже - удвоение всего спинного мозга.
Здравствуйте мамы) Меня зовут Юлия, в этом году на свет появилась моя дочка Мелисса, наша история началась так: Экстренное кесарево, причина неизвестна сказали в роддоме, что перестраховка, на мой вопрос как моя малышка мне ответили-в полном порядке здоровая девочка у вас родилась! На вторые сутки Мелисса перестала дышать, ИВЛ вентиляция легких сама не дышит, под вопросом ставили пневмонию.. В итоге судороги на 5 дне, и слова реаниматолога что не знай будет жить или нет😐Помню было страшно жить с этим, каждый день молилась за ее здоровье. В итоге моя малышка оказалась борцом и начала дышать сама, выписали сначала нас на стационар, пролежали месяц в неврологии, ставили:Целебральную ишемию, незрелость легких, порок сердца, незрелость мозга, пережили желтушку, кисты и прочее... Сейчас нам 3 месяца а я не могу выдохнуть и успокоится, я думала что все закончится когда нас выпишут но нет... Сейчас невролог ставит диагноз: ВПР ЦНС, агенезия мозолистого тела, желудочки срослись и увеличены в размере, гидроцефалия (голова у нас нормальной формы и размера), ослабленный тонус мышц. Были у окулиста, нам сказали что не следит за игрушками и плохи факусирует взгляд для своего возраста, то есть наблюдают психо-моторное отставание, зрение говорят -4. Я хожу и спрашиваю к врачей чего ждать и чем помочь, делаем курс массажа, медикаменты (Диакабр, калия орорат и кавинтон+уколы) нам из за проблем с сердцем нельзя пока ультрафарез. На данный момент ждем обследование ЭЭВ сна для исключения симптомов эпилепсии(плечиком подергивает иногда) Врачи не чего не прогнозируют, говорят многие отказываются я и думать не хочу об этом и нет у меня в голове что мой ребенок особенный, она просто в моих глазах позже развивается и все (+ стационар месяц почти спали) Мелисса активная девочка, различает голоса родных, знает интонацию когда с ней ласково удвбается, агукает, голову держит неуверенно но поднимает и старается удержать лежа на животе, меня беспокоит что совсем не интересуется игрушками и не факусирует на чем то конкретном взгляд, ноудио разглядывает все. Мне лишь повторяют что все что можно все делаем только ждать с там что Бог дал, страшно жить с этим, это мой первый ребенок, мне 20 лет, сложно понять что будет я много занимаюсь с ней и стараюсь не вешать нос. Но не верю что малышка особенная, все жду что вот все наладится.. С сердцем к нас хорошие улучшения порок проходит сам постепенно.. Подскажите кто знает мамочек с похожим диагнозом и как вы лечились? Чего ждать? И на что обращать внимание? Может ли все таки это пройти с возрастом? Читала всех, и хочу сказать, что молодцы все кто держится и не унывает, все думаю в наших руках, нам нельзя отчаиваться поэтому держимся до последнего и не сдаемся, терпения дорогие мамы
Септооптическая дисплазия (синдром де Морсье) - это порок развития переднего отдела головного мозга, который развивается ближе к концу первого месяца гестации и включает гипоплазию оптического нерва, отсутствие перегородки между передними отделами двух боковых желудочков, недостаточность гормонов гипофиза. Несмотря на то что причины могут быть множественными, у некоторых пациентов с септооптической дисплазией были обнаружены аномалии одного определенного гена (HESX1).
Симптомы могут включать снижение остроты зрения одного или обоих глаз, нистагм, косоглазие и эндокринную дисфункцию (включая дефицит гормона роста, гипотиреоз, надпочечниковую недостаточность, несахарный диабет и гипогонадизм). Могут развиваться судороги. Несмотря на то что у некоторых детей интеллект нормальный, у других отмечаются нарушения обучения, задержка умственного развития, детский церебральный паралич или другие виды задержки развития. Диагноз основывается на МРТ. Всех детей при выявлении этого заболевания необходимо обследовать на эндокринные дисфункции и нарушения развития. Лечение поддерживающее.
Анэнцефалия
Анэнцефалия - это отсутствие полушарий головного мозга. Отсутствующий головной мозг иногда замещается неправильно сформированной кистозной нервной тканью, которая может быть обнаженной или закрытой кожей. Части ствола мозга или спинного мозга могут отсутствовать или быть сформированными неправильно. Ребенок рождается мертвым или умирает в течение нескольких дней или недель. Лечение поддерживающее.
Энцефалоцеле
Энцефалоцеле это выпячивание нервной ткани и мозговых оболочек через дефект в черепе. Формирование дефекта связано с неполным закрытием свода черепа (cranium bifidum). Энцефалоцеле обычно возникают по средней линии и выпячивание происходит в любом месте от затылка до носовых отверстий, однако может быть асимметричным в лобной и теменной областях. Небольшое выпячивание может напоминать кефалогематому, однако на рентгенограмме заметен дефект черепа в его основании. Часто при энцефалоцеле отмечается гидроцефалия. Примерно у 50 % детей отмечаются другие врожденные аномалии.
Прогноз, который зависит от локализации и размера выпячивания, обычно хороший. В большинстве случаев энцефалоцеле можно успешно прооперировать. Даже при большом размере выпячивание, как правило, содержит преимущественно эктопированную нервную ткань, которую можно удалить без ухудшения функциональных возможностей. Если энцефалоцеле сочетается с другими тяжелыми аномалиями развития, решение о проведении операции может быть более трудным.
Неправильно сформированные полушария головного мозга
Полушария головного мозга могут быть большими, маленькими или асимметричными; извилины могут отсутствовать, быть необычно увеличенными или многочисленными и маленькими; при микроскопическом исследовании выглядящего нормальным головного мозга может обнаруживаться дезорганизация нормального расположения нейронов. Микроцефалия, умеренная или тяжелая задержка моторного и умственного развития и эпилепсия часто отмечаются при этих дефектах. Лечение поддерживающее, включает противосудорожные препараты при необходимости для контроля судорожного синдрома.
Голопрозэнцефалия
Голопрозэнцефалия развивается, когда в эмбриональном прозэнцефалоне (переднем мозге) не происходит сегментации и расхождения. Передний средний мозг, череп и лицо сформированы неправильно. Этот порок развития может вызываться дефектами гена sonic hedgehog («звуковой еж»). При тяжелом поражении плод может погибнуть до рождения. Лечение поддерживающее.
Лиссэнцефалия
Лиссэнцефалия состоит из аномально утолщенной коры, сниженной или отсутствующей дифференцировки на слои и диффузной гетеротопии нейронов. Причиной является нарушение миграции нейронов, процесса, с помощью которого незрелые нейроны соединяются с радиальной глией и перемещаются из места закладки около желудочков к поверхности головного мозга. Несколько дефектов отдельных генов могут быть причиной этого порока развития (например, LIS1). У детей при этом пороке развития отмечаются задержка умственного развития, мышечные спазмы и судороги. Лечение поддерживающее, однако многие дети умирают до двухлетнего возраста.
Полимикрогирия
Полимикрогирия, при которой извилины маленькие и имеются в большом количестве, считается следствием повреждения головного мозга между 17-й и 26-й неделями гестации. При этом могут отмечаться задержка умственного развития и судороги. Лечение поддерживающее.
Порэнцефалия
Порэнцефалия - это киста или полость в полушарии головного мозга, которая сообщается с желудочком. Может развиваться пре и постнатально. Этот дефект может вызываться пороком развития, воспалительным процессом или сосудистым осложнением, например внутрижелудочковым кровоизлиянием с распространением в паренхиму. Результаты неврологического обследования, как правило, отличаются от нормальных. Диагноз подтверждают при проведении КТ, МРТ или УЗИ головного мозга. Редко при порэнцефалии возникает прогрессирующая гидроцефалия. Прогноз различен; у небольшого числа пациентов развиваются только минимальные признаки со стороны нервной системы, у них отмечается нормальный интеллект. Лечение поддерживающее.
Гидранэнцефалия
Гидранэнцефалия - это крайняя форма порэнцефалии, при которой полушария головного мозга практически полностью отсутствуют. Обычно мозжечок и ствол мозга сформированы нормально, базальные ганглии интактны. Мозговые оболочки, кости и кожа над сводом черепа нормальные. Часто гидранэнцефалия диагностируется пренатально по УЗИ. Результаты неврологического обследования, как правило, отличаются от нормальных, ребенок развивается с нарушениями. Снаружи голова может выглядеть нормальной, однако при диафаноскопии свет просвечивает полностью. КТ или УЗИ подтверждают диагноз. Лечение поддерживающее, с шунтированием при избыточном нарастании размеров головы.
Шизенцефалия
Шизенцефалия, которая может быть классифицирована как одна из форм порэнцефалии, является следствием формирования аномальных извилин или расщелин в полушариях головного мозга. В отличие от порэнцефалии, которая, как считают, является следствием повреждения головного мозга, думают, что шизенцефалия представляет собой дефект миграции нейронов и, таким образом, истинный порок развития. Лечение поддерживающее.







